



This blog is written by Michael Gladstone, Principal at Atlas, as part of the From The Trenches feature of LifeSciVC.
Charles de Gaulle once asked, “How can you govern a country which has 246 varieties of cheese?” But that might seem like child’s play compared to the task of successfully drugging an immune system with >100,000 varieties of natural killer cell.
Immunology is divisive. New discoveries reveal, as Mike Gilman put it, “finer and finer subdivisions of cell types, ILs, and CDs.” This precision and complexity provide us many drug-target candidates, but they also deeply complicate the task of prioritizing among them.
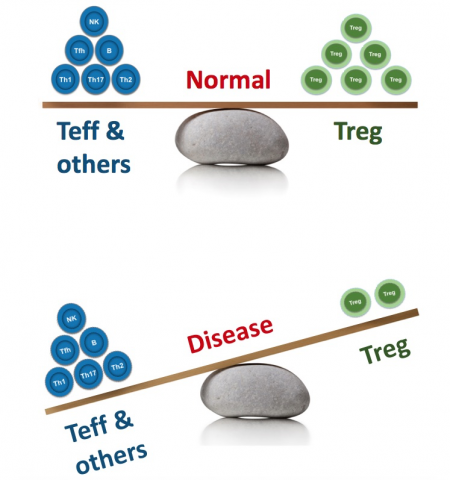
But on rare occasions, the combination of human genetics and clinical pharmacology can point us definitively in a new direction. This post is about one very specific cell type, the regulatory T cell (“Treg”). While comprising only ~2% of circulating lymphocytes, Tregs play an outsized, starring role in immune regulation.
Atlas and Sofinnova Partners closed a $35M Series A investment in a new company, Delinia, to develop drugs that directly potentiate and expand Tregs in vivo. Delinia’s therapies may truly re-induce self-tolerance and re-balance immunity in patients with autoimmune disease, rather than bluntly suppress immunity like today’s anti-inflammatories. I am obviously not at all impartial, as my employer is a co-lead investor and I am a board observer … but I’ll still say that I’m extremely excited about it.
Immune Homeostasis and Self-Tolerance
If the immune system is the body’s police force, who polices the police? Gershon and Kondo first identified immunity’s internal affairs department in 1970. They characterized a mysterious immune-regulatory T cell population, showing for the first time that T cells could actually moderate, as well as augment, immunity. Shortly thereafter, other groups showed that removing a mouse’s thymus initiated multi-organ inflammation and tissue damage, which could be rescued by provision of T cells from healthy animals.
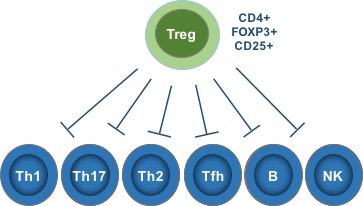
These experiments conclusively demonstrated the existence of a T cell population essential for maintaining self-tolerance. Researchers searched diligently for a way to identify these cells until, finally, in 1995 Sakaguchi et al famously identified CD25 as a selective marker of these cells. Scientists then identified the transcription factor FoxP3 as another essential marker, and these cells are now known as CD4+ CD25+ FoxP3+ regulatory T cells.
Tregs are responsible for maintaining tolerance to self-antigens and preventing rampant autoimmunity without restraining protective immunity against pathogens.
Focusing on Tregs
One fast way to appreciate anything’s importance is to see what happens when it’s broken. The pipe encased in my kitchen wall gave me this opportunity last winter, and a rare genetic disease called “IPEX” grants immunologists this privilege for Tregs.
Bruce has written about how “extreme clinical phenotypes” caused by rare genetic variations can provide essential target validation to drug discoverers (here). IPEX is a classic example: it is a severe multi-organ autoimmune disease caused by Treg dysfunction resulting from loss-of-function mutations in CD25 or FoxP3.
In IPEX patients, multiple immune pathways (e.g., Th1, Th2, Th17, B cells, monocytes, eosinophils, etc.) are highly dysregulated, and autoimmunity manifests in numerous tissues and organs (skin, lungs, gut, thyroid, pancreas, endocrine glands, and more). This illustrates two important aspects of the role normally played by Tregs:
- Tregs broadly regulate numerous immune activities and cell types
- Tregs are indispensable for self-tolerance across numerous organs and tissues
The biological implications of the extreme IPEX phenotype are supported by more common (but less disastrous) CD25 polymorphisms, which have been linked to numerous immune disorders. This is further supported by functional associations, as we see consistent evidence of suboptimal Treg numbers and/or activity in myriad autoimmune diseases.
CD25 and IL-2: Everything Old is New Again
Immunology-savvy readers may have noticed that I casually mentioned CD25 (which is the IL-2 receptor α-chain) as a selective Treg marker without acknowledging how surprising or paradoxical that might seem. When Robert Gallo et al discovered IL-2 in 1976, they literally called it “T cell growth factor” (TCGF), and subsequent studies revealed it to be a very potent immune stimulator. So potent that the recombinant cytokine was developed and FDA-approved as an immunotherapy for melanoma and renal cell carcinoma. This raises an obvious question: How can so general an immune stimulator as IL-2 also be an indispensable mediator of immune regulation and tolerance?
A Tale of Two IL-2’s
As any drug developer knows, dose matters. With IL-2, dose really matters, especially because we actually have two different types of IL-2 receptor. As shown below, there is both a high-affinity receptor (IL-2Rαβγ) and a low-affinity receptor (IL-2Rβγ). The former is selectively expressed at high levels on Tregs, while the latter is expressed very broadly throughout the immune system.

This explains why high-dose IL-2 is a potent yet toxic immune stimulator. With the large doses used by oncologists, blood IL-2 concentrations are high enough to stimulate both the low- and high-affinity IL-2 receptors. Due to the former’s far, far broader expression, the net effect here is very robust immune activation. While this produces striking remissions for some patients, it also provokes substantial toxicity, ranging from severe flu-like symptoms, vomiting, and diarrhea to vascular leak syndrome, whose manifestations include frequently life-threatening pulmonary edema. This is reminiscent in some ways of the cytokine release syndrome that frequently accompanies CAR-T therapy.
But might there be a way of zeroing in on that Treg-restricted high-affinity receptor to elicit immune-regulation, rather than immune-activation?
IL-2 Agonism: Bench to Bedside
A few ingenious hematology-oncology clinicians (including the Dana Farber’s Dr. Jerry Ritz, Dr. Robert Soiffer, and Dr. John Koreth) had a great idea: What if we could selectively stimulate the high affinity-Treg IL-2Rαβγ, without stimulating the broadly-expressed low-affinity receptor? If so, could we selectively augment Tregs without activating the rest of the leukocyte compartment and use this approach to treat autoimmunity?
These Dana Farber transplant physicians treat graft vs. host disease (GVHD), a frequently severe autoimmune disease resulting from a stem cell donor’s immune system attacking the recipient’s tissues. GVHD has high rates of morbidity and mortality, and it is among the most severe and intractable manifestations of autoimmune disease.
Unfortunately, no truly IL-2Rαβγ-selective agonist existed at the time, so the Dana Farber team had to MacGyver a cruder approach. They knew IL-2 itself activated both receptors, but they thought maybe they could use frequent, very low IL-2 doses to repeatedly tickle the high-affinity IL-2Rαβγ on Tregs, without over-stimulating the rest of the immune system bearing the low-affinity IL-2R. These physicians walked a very narrow line: too low or too infrequent an IL-2 dose, and they wouldn’t stimulate Tregs; too much or too frequent an IL-2 dose, and they may further stimulate an already overactive immune system in patients with severe autoimmunity, compromising safety and efficacy.
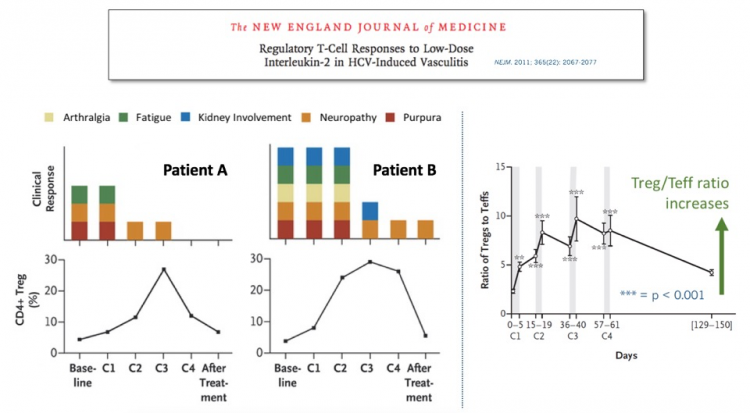
After some careful clinical empiricism, the Farber group identified an IL-2 regimen that safely elicited a modest increase in Tregs, which produced striking clinical benefit in some patients with severe, steroid-refractory GVHD. These impressive results were reported in the New England Journal of Medicine.
But this was admittedly an exploratory single-center trial in one clinical indication. Do we really know that IL-2Rαβγ agonism can activate Tregs in other settings, or that pharmacological Treg activation will have clinical benefit anywhere else?
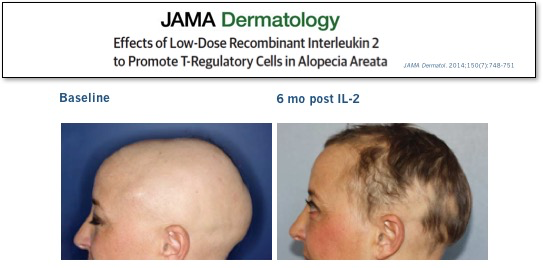
We didn’t have to wait long for answers to these questions. In the same NEJM issue, a group on another continent treating a very different severe autoimmune disease reported similarly impressive results in patients with refractory HCV-induced vasculitis. Subsequent high-profile peer-reviewed clinical results with low-dose IL-2 in other diseases, ranging from lupus to allopecia, have further validated this approach.
In these clinical studies, even modest Treg increases have been associated with clinical improvement. And, very importantly, these Treg increases have not produced increased infection risk, unlike many potent anti-inflammatories used to treat these diseases (e.g., TNF inhibitors, IL-17 inhibitors, steroids, calcineurin inhibitors, JAK inhibitors, etc. ad infinitum). This makes sense, given that Tregs naturally are tasked with controlling autoimmunity without restraining protective anti-pathogen immunity. But it’s still great to see that the clinical experience so far is supportive of this hypothesis.
Bedside Back to the Bench
So if Tregs can be pharmacologically augmented (albeit crudely) and this can benefit patients with a variety of autoimmune conditions, why hasn’t low-dose IL-2 replaced all of the sledgehammer immune-suppressor drugs?
While low-dose IL-2 was essential for pharmacologically validating the clinical utility and apparent safety of Treg potentiation, two major liabilities prevent its optimal and broad deployment in the real world.
- IL-2 is only slightly more potent on the “high-affinity” Treg IL-2Rαβγ vs the “low-affinity” IL-2Rβγ. IL-2 grants us only a very narrow window between activating Tregs (which we want) and activating effector T cells, CD8 T cells, NK cells, monocytes, eosinophils, etc. (which we definitely don’t want)
- IL-2 has a very short elimination half-life (1-2 hours), which requires frequent injection dosing and results in sub-optimal Treg stimulation. It also further complicates the very narrow therapeutic window we face, given point #1 above
The consequences of this keep IL-2 from being a broadly viable therapy for immune disorders:
- Suboptimal efficacy: it is literally impossible to use a high enough IL-2 dose to maximally augment Tregs without also stimulating other immune cells (which entails toxicity and is counterproductive for efficacy)
- Frequent (often daily) injections are required
- Poor tolerability: again, thanks to IL-2’s low degree of high- vs. low-affinity receptor selectivity, even the very small doses of IL-2 used in these studies stimulate the low-affinity IL-2R enough to produce problematic constitutional symptoms for many patients, necessitating even further dose reduction
Enter Delinia
The clinicians who pioneered these proof-of-concept studies for low-dose IL-2 deserve immense credit for their ingenuity. These folks are brilliant, and they did something great (which is why Dr. Ritz is among the Treg/immunology luminaries advising us on Delinia’s SAB).
Likewise, I deserve a ton of credit for that time I replaced my house’s disgusting inlet water filter using broken pliers, rather than the elegant made-for-purpose wrench that I didn’t know I could buy for $7 at the hardware store. But I found that wrench and bought it, and now replacing my water filter is easy and fast, and I don’t spill freezing sediment-rich water all over my shoes. (Wrench pictured here.)
My hope is that Delinia has invented the wrench that will allow us to retire low-dose IL-2, which is a useful and important contraption, but not the best medicine to potently, safely unlock this biology. Here’s what we need from an ideal IL-2Rαβγ agonist:
- Very potent activator of high-affinity IL-2Rαβγ to potently stimulate Treg expansion and activity
- Undetectable binding/activation of low-affinity IL-2Rβγ to avoid stimulating efficacy-opposing and toxicity-inducing activity on the broader immune compartment. This selectivity is paramount; the whole therapeutic hypothesis depends on maximizing the window between stimulation of the high- and low-affinity IL-2 receptors
- Enhanced PK both to optimize the duration of target engagement to increase potency and to limit the frequency of dosing
Delinia’s founding team was a perfect pairing of individuals to tackle this problem, and I think they have cracked it with their proprietary novel recombinant biologics that potently and selectively activate IL-2Rαβγ (more details to be revealed in the future…)
Jeff Greve, founder and CSO, is an immunology-savvy protein engineer who previously led R&D programs at both Big Pharma (Bayer) and biotech (Exelixis, aTyr). He has lots of experience working on IL-2 immunobiology and biochemistry, which positioned him well to invent, validate, and optimize Delinia’s unique approach.
Mike Rosenblum is an MD, PhD physician-scientist and Assistant Professor at UCSF. His clinical practice and laboratory research are both focused on severe autoimmune diseases affecting the skin, and on therapeutic approaches (especially Treg augmentation) to re-establish immune tolerance. He’s also a lead investigator on a number of UCSF’s clinical trials using adoptively transferred Tregs to treat autoimmune diseases.
On the back of Jeff and Mike’s vision and initial data, Atlas and co-lead investor Sofinnova Partners seeded Delinia last year to optimize several of Delinia’s lead molecules, generate additional validating data, and expand their already strong IP footprint. The team executed wonderfully on that plan, so Atlas and Sofinnova moved forward with a $35M Series A.
We were all incredibly fortunate that Saurabh Saha agreed to join the company this year as CEO. Saurabh is a Venture Partner and Entrepreneur-in-Residence at Atlas, where he’s helped evaluate countless investment opportunities while also serving, until recently, as Chief Medical Officer at Atlas portfolio company Synlogic. Saurabh is a wizard when it comes to quarterbacking efficient and creative translational and clinical development, skills that we hope will serve us all well as we embark upon the development of unique therapies with potential across a broad array of autoimmune, inflammatory, and fibrotic diseases.
Looking Ahead
I’m a big believer in the IO revolution. One concept I love about checkpoint inhibitors is that they enable personalization without customization. Off-the-shelf PD-1 blockade enables a patient’s unique immune system to amplify its attack on that patient’s unique tumor, recognizing a variety of antigens and using a variety of approaches (cytotoxicity, secreting numerous cytokines and chemokines, etc.). I like not having to guess which mutations are most important to target, or what cytokines are most important to use. The immune system is much smarter than I am (after all, it’s the one that knows what to do with 100,000 kinds of NK cell), so I’d rather give it the proper pharmacological encouragement and let it sort that all out on its own.
Unfortunately, we haven’t really been able to do that yet for autoimmune disease. We acknowledge that autoimmunity results from failed immune tolerance to auto-antigens. This is of tautological obviousness. Yet we don’t treat autoimmunity by restoring tolerance; we treat it by selecting an immune cell to deplete or a central pathway or cytokine to block. But these cells and pathways are often highly redundant (which limits efficacy) and also central for protecting us from pathogens (which is why these drugs increase infection risk).
Many of these drugs have made a huge difference in the lives of millions of patients. We certainly can’t overlook that. But autoimmunity continues to inflict tremendous suffering, and I wonder if we might be asymptotically approaching the ceiling of the risk/benefit ratio for immune suppressors.
What excites me about Delinia is the opportunity to borrow a page from the immuno-oncology playbook, by empowering the adaptive immune system to do the hard work for us. For the first time, we may be able to pharmacologically augment the exact biological pathway that has evolved for millennia to maintain self-tolerance and immune homeostasis. Delinia allows us to go upstream by targeting a cell type that naturally regulates immunity in many tissues, using many mechanisms, and yet does so while leaving intact our anti-pathogen defenses. And just like PD-1 inhibitors have benefitted patients with a multitude of tumor types, I think Treg activation has the potential to serve as the new bedrock for treating a very wide range of autoimmune diseases.
Given what low-dose IL-2 has accomplished despite its drawbacks, we’re very excited to see what the Delinia team can do with their therapies.