



The IQVIA Institute released a data-rich report in April 2019 on the state of clinical R&D in the US pharmaceutical industry titled “The Changing Landscape of Research and Development: Innovation, Drivers of Change, and Evolution of Clinical Trial Productivity”. The report is a fascinating deep-dive into lots of data cuts around what’s going on.
The increasing role of biotech in marketing their own drugs was the subject of much of the coverage on the report (here, here), which is indeed an interesting trend to see. But rather than opine on that observation or the full scope of the report, I wanted to highlight a number of findings, largely glossed over elsewhere, relevant to the state of clinical trial activity and approvals that are relevant to biotech investors.
With clinical trial activity in the industry up 35% over the past few years, with over 4700 trials starting in 2018, it’s worth looking at some of the clinical trial metrics, especially since the features of clinical trials, such as the number of arms, numbers of patients, and years of trial follow-up are major drivers of costs.
Before diving in, some context on the data in the report: IQVIA tracks all the New Active Substances (NASs) that were launched in the US in 2018 (irrespective of what year they were approved). This includes 59 NASs for 2018, roughly half of which were orphan disease drugs. Here are a few of the key findings as they relate to clinical trial activity.
Features of the clinical trials in the regulatory approval packages:
- Randomized controlled trials (RCTs) clearly remain the gold standard for approval: nearly 90% of drugs included RCTs in their regulatory packages.
- Trials with active control arms are more common today, and were included in the regulatory filings of nearly half of these new drugs (46%), versus only 20% a few years ago. This highlights the increasing importance of comparative effectiveness in disease indications where existing standards of care are in place.
- Only one registrational trial was required in over 40% (25/59) of these approvals; so much for the old “two well-controlled Phase 3 trials are required” regulatory guidance of the past.
- Only Phase 1 or 2 studies did the trick in 12% (7) of the drugs’ approvals; these had no Phase 3 studies, so are obviously special situations.
Magnitude of patient exposures and time.
- There’s a huge range in the number of patients in these regulatory filings, as captured by the distribution below which was derived from the report. Roughly half of the approvals had fewer than 500 patients treated.
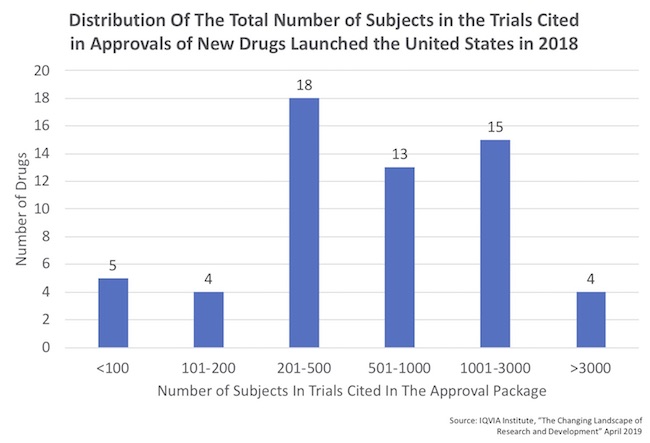
- On the small end of the distribution, five drugs were approved with fewer than 100 patients of data. All were, as expected, orphan drugs. Four of these were approved on the basis on only a single trial. The 5th was elapegademase for ADA-SCID, which was approved on the basis of only 10 patients in two registrational trials!
- The average number of patient-years of exposure supporting new drug launches between 2016-2018 has been 1800-3800 patient-years. Patient-years of exposure is often an important metric for understanding safety in particular, but also durability of efficacy
- Orphan drugs had, on average, around 1000 patient-years of therapy over that period. While smaller than the non-orphan drugs, as expected, it’s not as small as I might have thought (e.g., could be 200 patients followed for 5-years of treatment or 5-years post-cell/gene therapy)
- Collectively, achieving this magnitude of patient exposures has required a significant amount of development time: the cumulative years in the clinic and registration remains long and hasn’t changed as the average in recent years is approximately 12+ years. This rough average time holds for both orphan drugs and non-orphans, interestingly.
- Although registrational trials for orphan drugs often include far fewer numbers of patients (about 5-fold fewer, ~430 vs ~2300 patients enrolled), these R&D programs still typically require long durations: in fact, the average trial duration for orphan drugs is actually 12% longer than for non-orphans, based on the 4-year averages (7.6 years vs 6.7 years). This was a surprising finding, and presumably reflects the broad range of orphan indications beyond just rare monogenic diseases.
These metrics highlight important aspects of the current clinical landscape. Integrating these with our experience as early stage investors, and a few key takeaways emerge:
- Active comparators with RCTs, even for orphan drugs in crowded classes, will likely be even more common going forward. Understanding comparability versus standard of care as early in development as practical will be increasingly important. Trying to run trials without them to save capital is not a recommended approach; plan the right study and convince investors to fund it, rather than cutting corners.
- Single arm trials, poorly controlled, will continue to face challenges – as we all know from history. In cancer studies, this often leads to false positives, and thus late stage failures over time. When they are used in rare orphan diseases, there are frequently no relevant long-term natural histories to rely on, creating further concerns. In these settings, companies should invest early (while in preclinical) to set up observational clinical studies aimed at creating adequate historic controls and benchmarks.
- Going forward, we should expect that drug approvals will likely require ~1000 patient-years of exposure for “mainstream” orphan indications, and at least 2-3x more than that in non-orphan drugs. Gene and cell therapies with dramatic effects can be expected to have shorter aggregate exposures, but benefit from a constant accrual of patient-years even after a one-and-done intervention.
- While special situations exist, unless there’s dramatic efficacy in early trials, very few drugs have or will be able to jump from Phase 1 or 2 straight to approval. It’s worth skeptically pressure-testing any pitches from teams that claim this expedited straight-to-approval approach is their base case plan.
- Orphan drugs, while more efficient in some ways, are not dramatically faster in clinical development than non-orphan programs, due to the need to accumulate patient-years of safety and efficacy data from smaller overall patient numbers.
Thanks to the team at the IQVIA Institute for a stimulating and thought-provoking read.