



By Cody Tranbarger, EIR at Atlas Venture, as part of the From The Trenches feature of LifeSciVC
For decades, muscular dystrophy has stubbornly persisted among the most intractable categories of human disease. Despite recent progress with AAV Gene Therapy and Antibody-Oligonucleotide Conjugates, a broad range of muscular dystrophies are still untreatable, and collectively represent an enormous unmet medical need. At the core of these disorders lies the Dystrophin-Glycoprotein Complex (DGC) – a massive, multi-protein structure responsible for anchoring muscle cells to their surrounding environment. While the DGC’s functional importance was understood as early as the 1980s, a unified picture of its molecular architecture has remained elusive, hampering efforts to develop truly targeted therapies.
That is, until now. Two recent studies, published in tandem by Nature (Liu et al. (2024), Wan et al. (2024)), have finally unveiled near-atomic resolution structures of the native, fully intact DGC, offering profound insights that not both redefine our understanding of the complex itself and open new therapeutic frontiers in muscular dystrophy. Together, they’re a tour de force of structural biology and, in my view, haven’t received nearly the attention they deserve. At the risk of diving into the scientific weeds, what follows is an attempt to highlight the importance of these findings and their implications for drug development in muscular dystrophy.
Looking Back: Piecing Together a Patchwork DGC
The DGC is a large, multi-protein assembly that plays a critical role in maintaining muscle integrity by linking the intracellular actin cytoskeleton to the extracellular matrix (ECM). This structural connection acts as a molecular shock absorber, facilitating lateral and vertical force transmission while protecting muscle fibers from contraction-induced damage. The DGC consists of three major subcomplexes (with frustratingly similar names): the dystrophin-associated cytoskeletal complex, which includes dystrophin and α-dystrobrevin that anchor actin to the membrane; the dystroglycan subcomplex, composed of α- and β-dystroglycan, which bridges dystrophin to ECM proteins like laminin-211; and the sarcoglycan-sarcospan subcomplex, containing α-, β-, γ-, and δ-sarcoglycans, as well as sarcospan, which collectively stabilize the complex within the sarcolemma (muscle cell membrane). Genetic loss-of-function variants in many DGC components compromise the mechanical stability of the sarcolemma, resulting in various muscular dystrophies, such as Duchenne Muscular Dystrophy (DMD), Limb-Girdle Muscular Dystrophy (LGMD), and Congenital Muscular Dystrophy (CMD). As such, the DGC and its underlying biophysical and mechanical properties have long been of high interest for academics and drug developers alike. 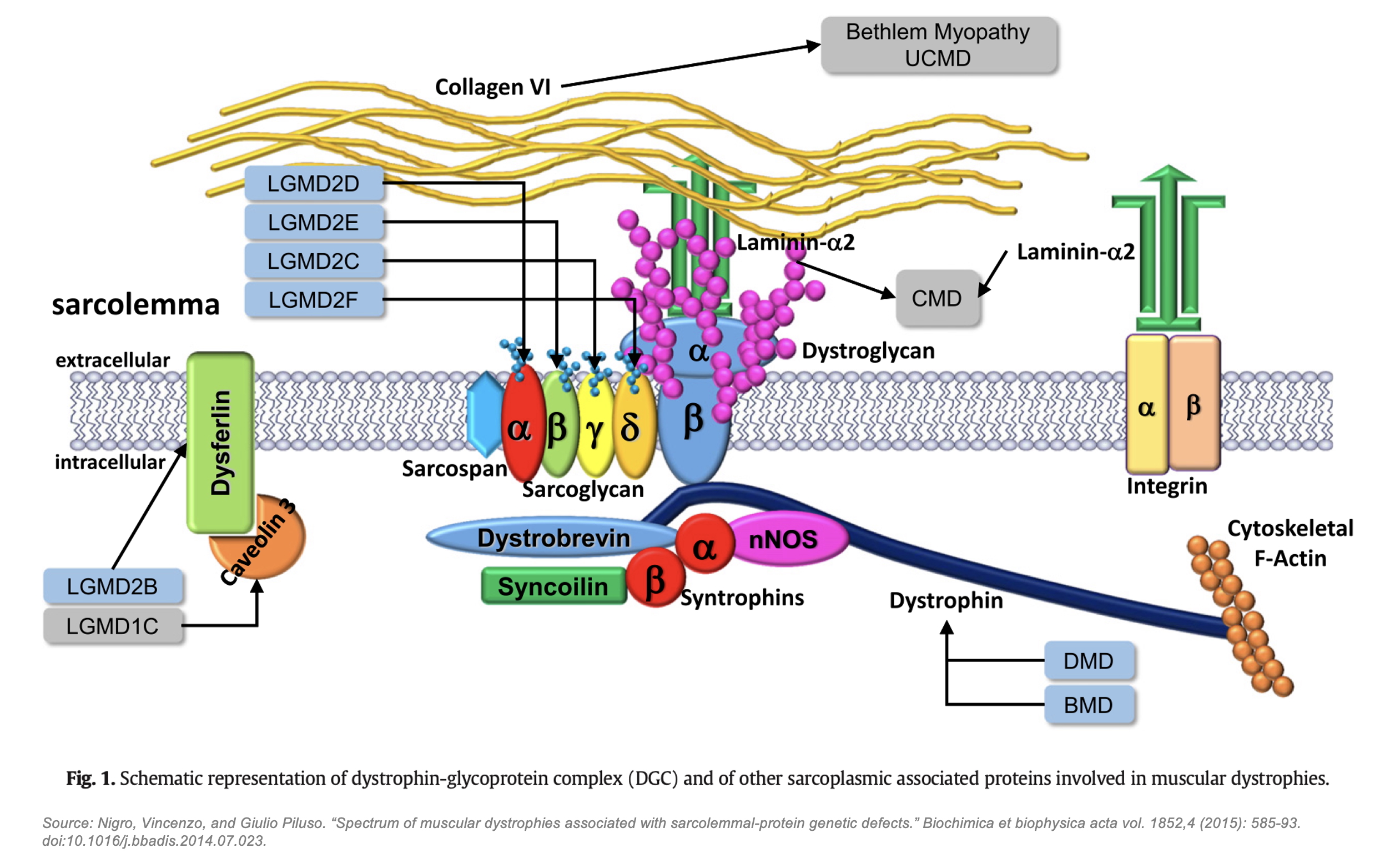
Despite decades of study, a complete structural model of the DGC remained out of reach. Early work throughout the 1990s and early 2000s relied on co-immunoprecipitation, domain-specific crystallography, and nuclear magnetic resonance to analyze individual DGC components, such as dystrophin’s WW and ZZ domains, dystroglycan’s ECM-binding glycosylation residues, individual sarcoglycan subunits, and the cytoplasmic syntrophins and α-dystrobrevin. These methods, however, were incapable of capturing the full multi-protein assembly, leading to a patchwork of assumptions that have until now comprised the field’s unified view of the DGC, such as:
- Dystroglycan has been assumed to be the core anchor of the DGC.
- Sarcoglycans have been thought to form an independent subcomplex at the periphery of the DGC.
- Sarcospan has been believed to play a minor, auxiliary role in stabilizing the sarcoglycan complex.
- Dystrophin’s binding interactions have been only partially deciphered, and the WW domain’s binding to β-dystroglycan has been thought to be the dominant interaction.
This incomplete picture of the DGC has constrained the development of new therapeutic strategies in muscular dystrophy. Without a fully resolved structure, efforts to design targeted interventions have been hampered by uncertainty regarding which domains are essential for function, how subunits interact dynamically, and how disease-causing mutations disrupt DGC assembly. As a result, most therapeutic mechanisms directed toward DGC restoration / stabilization, such as utrophin and GALGT2, have failed; and even those that have succeeded in getting to FDA approval, such as micro-dystrophin and exon skipping, have left much to be desired in terms of actual functional benefits for patients.
Breaking Through: Cryo-EM Cracks the DGC
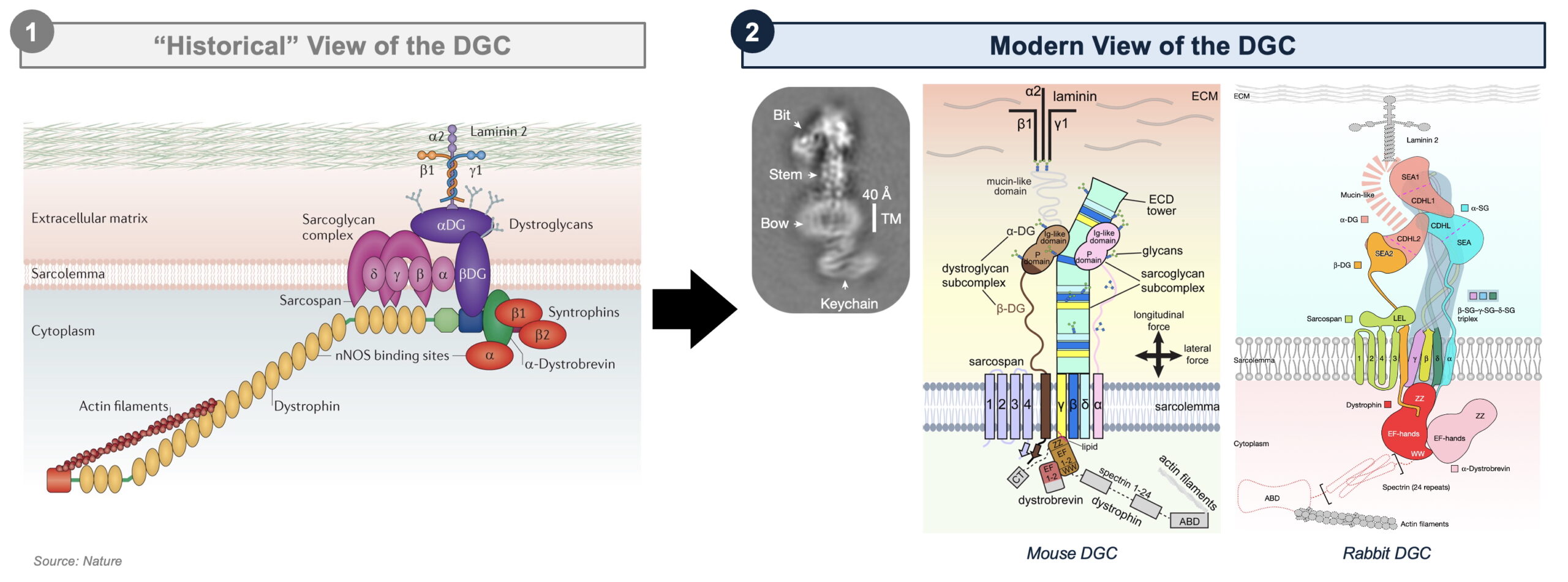
Enter Cryo-Electron Microscopy (Cryo-EM). Unlike traditional X-ray crystallography, which requires the protein of interest to form stable crystals, Cryo-EM enables the visualization of large, flexible protein assemblies in their native states. Since its emergence in the early 2010s, Cryo-EM has revolutionized structural biology, enabling breakthroughs in our understanding of a broad range of complex macromolecular assemblies, including the ribosome, the spliceosome, ion channels, viral capsids; and now, the DGC. Liu et al. and Wan et al. beautifully integrate single-particle Cryo-EM and computational modeling to achieve the first high resolution 3D reconstruction of the intact DGC from mouse and rabbit skeletal muscle at an impressive 3.5–4.3 Å resolution.
Together, these studies offer several new revelations; among the most significant are:
- A new structural core: rather than their previously assumed peripheral location, β-, γ-, and δ-sarcoglycans form a central scaffold – an extracellular “tower” comprised of a triple β-helix – that stabilizes dystroglycan and dystrophin.
- A critical, and precedented, curvature: due to specific amino acid variations, this sarcoglycan scaffold assembles with a bent curvature, which may ensure sarcolemma stability during contraction-induced stress. Notably, this feature has parallels to certain pathogens, which also utilize bent β-helices to enhance surface interactions and mechanical resilience, suggesting some degree of evolutionary conservation.
- A more promiscuous Dystrophin: Rather than binding only to β-dystroglycan, dystrophin’s WW and ZZ domain also interact with sarcoglycans, α-dystrobrevin, and lipids, revealing a more complex web of interactions that regulate DGC integrity.
- A revised role for Sarcospan: Rather than directly binding α-dystroglycan, sarcospan stabilizes β-dystroglycan by positioning it between sarcoglycans, clarifying its role as a structural anchor within the sarcolemma.
Not only does this work rewrite the textbook model of DGC assembly, but it also deepens our understanding of the molecular pathology of muscular dystrophy. Between the two studies, more than 110 pathogenic mutations were structurally rationalized, directly linking single-residue changes to DGC destabilization, and establishing a directional genotype-phenotype relationship between the extent of DGC dysfunction and disease severity across a variety of muscular dystrophies driven by loss-of-function in individual DGC components.
Looking Forward: From Structures to Medicines
The structural resolution of the DGC represents a milestone decades in the making; and these structural advances carry important implications, both for existing mechanisms and for future therapeutic avenues.
First, they provide guidance for the design and refinement of micro-dystrophin gene therapy constructs for DMD. By necessity, micro-dystrophins lack large regions of the wild-type protein, and there has been substantial debate over the years regarding optimal construct design. Prominent among these controversies has been the importance of retaining certain domains, such as the C-terminal coiled-coil region that was believed to mediate binding to α-dystrobrevin. These structures, however, propose and validate a newly discovered binding interaction between dystrophin’s WW domain and α-dystrobrevin, seemingly refuting the need for coiled-coil inclusion and further reinforcing the indispensability of the WW domain.
These findings also provide a roadmap for the development of sarcoglycan-directed small molecule correctors for LGMDs. As the authors methodically demonstrate, many LGMD missense mutations drive pathology by destabilizing the β-helix ECD tower. Therefore, if a small molecule chaperone could achieve partial correction of sarcoglycan misfolding and / or localization, it could restore DGC assembly and stability. Cystic Fibrosis is the obvious parallel, and in fact, CFTR correctors have demonstrated the ability to rescue LGMD2D due to α-sarcoglycan (SGCA) missense variants in vitro and in vivo – a promising starting point.
Finally, these new learnings allow us to look beyond the DGC with greater confidence by providing valuable insights into the molecular interfaces between individual DGC components and their ECM ligands. This newfound visibility allows for deeper interrogation of the dysfunctional DGC / ECM interactions that underlie several underserved subtypes of LGMD and CMD, and may open up new therapeutic opportunities for targeted biological approaches in the extracellular space.
While plenty of translational challenges remain, these studies have armed the field with a molecular framework to guide next-generation drug discovery efforts. If history is any indicator, a new wave of therapies may be on the horizon.
Here at Atlas Venture, we’re thinking deeply about all these new avenues and more. We’re proud to have played a role in advancing innovation for musculoskeletal disorders at Dyne and elsewhere, and we’re excited to continue building great companies that will bring great drugs to muscular dystrophy patients worldwide. Stay tuned.
References
Liu, Shiheng et al. “Native DGC structure rationalizes muscular dystrophy-causing mutations.” Nature vol. 637,8048 (2025): 1261-1271. doi:10.1038/s41586-024-08324-w
Wan, Li et al. “Structure and assembly of the dystrophin glycoprotein complex.” Nature vol. 637,8048 (2025): 1252-1260. doi:10.1038/s41586-024-08310-2