



This blog was written by Mike Gilman, Atlas’ EIR and CEO of Padlock Therapeutics, as part of the “From the Trenches” feature of LifeSciVC.
I thought I was done with immunology.
I’m not formally trained as an immunologist, but I fell for it hard back in graduate school, when immunology was leading the molecular biology revolution. Every student of modern biology should read the original 1976 Hozumi and Tonegawa paper in PNAS demonstrating immunoglobulin gene arrangement — even the Materials and Methods section is a gripping read. But over the decades, immunology and I, we grew apart. You see, I’m a lumper — I like simple ideas that unite and explain disparate facts (as the Tonegawa experiment elegantly explained how antibody diversity is generated) — but immunology has become a splitter. The last two or three decades of immunology have been about finer and finer subdivisions — of cell types, of ILs, of CDs. In drug development, that kind of precision can stop helping you. After all, most of our successful immunological drugs are pretty blunt instruments, taking out whole swaths of the immune system. Although elegance and selectivity are intellectually compelling, when it comes to an enemy like autoimmune disease you need heavy weapons. So how, I wondered, are contemporary developments in immunology going to help us create better medicines for patients?
And then I discovered the PADs.
Well, I didn’t discover them, of course — they’ve been known for decades — but what I learned in my little sabbatical from corporate science here at Atlas is that autoimmune disease is caused by autoantigens.
Duh, you say.
But that statement has more content than your typical tautology. Autoantigens are active participants in the initiation and development of autoimmunity. After all, breaking tolerance is not a trivial thing — we have elaborate mechanisms to make sure that it doesn’t happen. So you need to produce autoantigens in sufficient quantity and context to prompt your immune system into performing an unnatural act. Then, over subsequent years and decades, you have to continue to produce autoantigens to mature the immune response to the point that it becomes clinically relevant. Once disease gets going, autoantigens are the raw fuel for the inflammatory cycle that sets up in target organs, and they drive the formation and deposition of immune complexes, which account for much of the morbidity and mortality in patients.
So, yeah, autoantigens are kind of a big deal. What if you could stop the body from producing them? What if you could deprive the immune system of its inflammatory fuel? Better yet, what if you could prevent a tolerance break from maturing into disease altogether? And all of this without messing with the immune system itself. That would be pretty good, right?
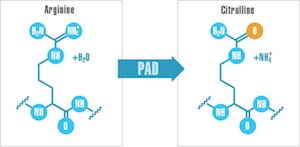 And that brings us back to the PADs, protein-arginine deiminases, an unassuming family of five enzymes that perform a simple post-translational modification of proteins, converting arginine side chains to the related amino acid citrulline. This is a pretty subtle modification, but it has a couple of potential consequences. One is that it removes a positive charge from the surface of the protein, which could impact structure and function. The other is that in people with certain genetic backgrounds, these citrullinated proteins are immunogenic — in other words, the PAD enzymes produce autoantigens.
And that brings us back to the PADs, protein-arginine deiminases, an unassuming family of five enzymes that perform a simple post-translational modification of proteins, converting arginine side chains to the related amino acid citrulline. This is a pretty subtle modification, but it has a couple of potential consequences. One is that it removes a positive charge from the surface of the protein, which could impact structure and function. The other is that in people with certain genetic backgrounds, these citrullinated proteins are immunogenic — in other words, the PAD enzymes produce autoantigens.
Another thing you need to know about PADs: when assayed in vitro, they require millimolar quantities of calcium for activity. That implies a couple of important things. Inside cells, where the Ca++ concentration is about 100 nM, PADs must be under some other form of regulation as yet unknown. And if PADs are released from cells and find themselves happily awash in the 2 mM Ca++ of the extracellular compartment, they will promiscuously citrullinate whatever unfortunate proteins they encounter.
PADs and rheumatoid arthritis
Antibodies specifically associated with rheumatoid arthritis have been recognized for over seventy years. Rheumatoid factor, an antibody directed against the Fc domain of IgG, was first associated with RA in 1940. A second RA-associated antibody, called anti-perinuclear factor, considerably more specific for RA than rheumatoid factor, was reported in 1964. In 1998, anti-perinuclear factor and other common RA-associated antibodies were shown to recognize citrullinated proteins. We now know that roughly 75% of RA patients have antibodies to citrullinated protein antigens (ACPA). Moreover, the presence of ACPA is up to 98% specific for RA. So if you have these antibodies, then you either have RA or you’re going to get it. Indeed, ACPA can be found in patients up to a decade before they show up in the doctor’s office with clinical symptoms of RA, suggesting that breaking tolerance to citrullinated proteins is an early step in the development of disease.
Interestingly, there are two principal risk factors for ACPA-positive RA. One is smoking and the other is a family of HLA alleles (the “shared epitope”) that encode Class II proteins that selectively bind citrullinated peptides for presentation to T cells. These observations have led to a model for the development of RA. In this model, RA begins with tissue injury at a distal site, for example in the airway epithelium of smokers. The injury causes cells to release PAD enzymes, which become ectopically activated and citrullinate proteins in the extracellular environment. In the context of the shared epitope, citrullinated peptides are presented to T cells and tolerance can be broken. Years go by, and as the patient continues to smoke and injure his airways, more PADs are released, more citrullinated antigens generated, and the immune response steadily matures and intensifies. Eventually, the immune response to citrullinated proteins reaches a threshold and, perhaps accelerated by trauma to a joint, the immune system sets up shop in the synovial compartment and clinically evident joint inflammation and destruction ensues.
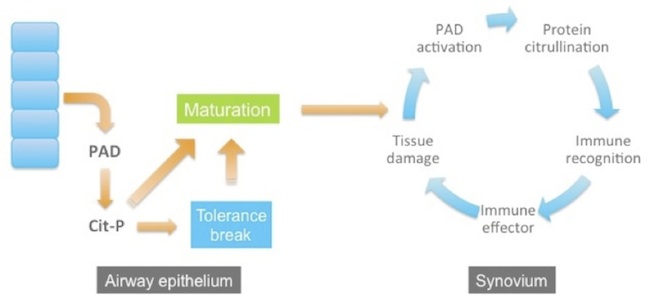
Now imagine you had a PAD inhibitor. You might be able to break the inflammatory cycle in patients with established disease by turning off antigen production. This idea is especially attractive for roughly 15% of RA patients, who make a fascinating autoantibody that allosterically activates PAD4. These patients run super-hot: their synovial compartment is chock-full of PAD activity, citrullinated proteins, ACPA, and B cells secreting these antibodies, and they have severely erosive disease. One of the complexities of autoimmune disease is epitope spreading — by the time patients present with active disease they are already making antibodies to many different antigens, so knocking out any single antigen is unlikely to make much of a difference. But these particular patients may have a relatively pure ACPA-driven disease, essentially turbocharged by PAD4 activity. In these folks, a PAD4 inhibitor may be just the ticket.
Perhaps even more exciting is the possibility of treating patients who have broken tolerance to citrullinated proteins but have not yet matured their immune response to the point where they’ve developed clinical disease. These patients are in principle pretty straightforward to find — there’s a commercial diagnostic for ACPA that is widely used to confirm an RA diagnosis today. Treating patients who are ACPA-positive but disease-free with a PAD inhibitor offers the possibility of delaying or even preventing the onset of RA entirely.
PADs and lupus
Have a look at this movie. Go ahead, it’s only twenty-five seconds long. The blue things bouncing around are primary human neutrophils with their weird lobulated nuclei. In this experiment, they are being exposed to serum from a lupus patient. After a while, some of the nuclei swell and expand and then — pow! — those green thermonuclear explosions are neutrophil extracellular traps (NETs). NETs are a form of ritual suicide performed by neutrophils in response to invading bacteria and fungi, part of their bag of antimicrobial tricks. NETs are sticky webs of decondensed chromatin studded with antimicrobial granule proteins and chemokines. They physically trap bugs, kill them, and summon macrophages to clean up the mess. NET formation requires citrullination of histones by PAD4; PAD4-null mice don’t make NETs.
Although NETs were only discovered about a decade ago, they have rapidly found their way into the etiology of many diseases. Perhaps the most intriguing connection is in lupus. If you look at the list of NET components, it reads like a who’s who of common autoantigens in lupus and other systemic autoimmune diseases — DNA, histones, other nuclear components, neutrophil cytoplasmic proteins. This observation has led to the hypothesis that NETs are the source of bulk autoantigen in lupus. NET-ing neutrophils dump large quantities of this material into the extracellular space and do so specifically at sites of infection, where the immune system is on high alert, TLRs fully engaged. It’s a perfect storm for tolerance break. In analogy to RA, this initial tolerance break is presumably subclinical; as a patient acquires subsequent infections, leading to more NETs, the autoimmune response matures and intensifies until clinical autoimmunity develops. We also know that lupus is a flaring disease and that flares are often associated with infections. The NET hypothesis explains why: each time a new infection arises, neutrophils are mobilized and produce NETs – and another log is tossed on the inflammatory fire.
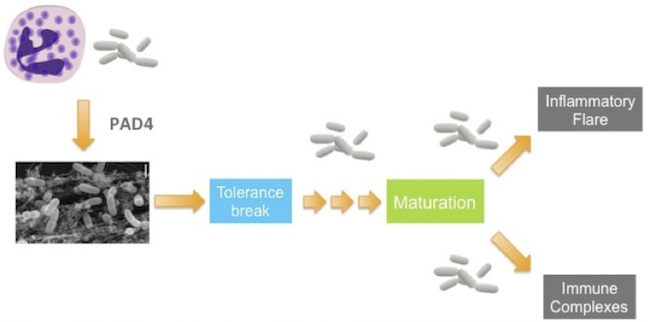
So in lupus, a PAD inhibitor — specifically a PAD4 inhibitor — could suppress NET formation and choke off the supply line for autoantigen, attenuating autoimmune inflammation, preventing flares and the formation and deposition of immune complexes, which are largely responsible for end-organ damage in lupus patients.
PADs and multiple sclerosis
PADs figure a bit differently in MS. And whether they are material here depends on where you stand on the outside-in versus the inside-out models for MS. The predominant model today — or at least the one favored by immunologists — is that MS is an outside-in disease. That is, the primary defect in MS is in the peripheral immune system — a legitimately-provoked immune response (e.g., to a virus) that unfortunately cross-reacts to a component of myelin, leading to an autoimmune attack on the CNS. The alternative model is that the primary injury is actually in the brain and that the CNS inflammation observed in many MS patients is a response to that injury. That the outside-in model is in vogue is evident from the list of approved drugs for MS, which are basically all immunomodulators. Although they are quite effective at preventing inflammatory lesions and clinical relapses, they do not have much impact on the underlying disability progression in these patients. Moreover, there are progressive forms of MS in which demyelination and disability progress inexorably in the absence of any obvious inflammation. And so far progressive MS has not responded to available immunomodulatory drugs. So it does seem reasonable to think that there’s something else going on in the brains of MS patients.
What might that be? Well, it’s been known for some time that myelin components, particularly myelin basic protein (MBP), are differentially modified in MS brain relative to healthy controls. Among those modifications is citrullination. Roughly 45% of MBP is citrullinated in the brains of MS patients, 90% in patients with Marburg variant, which is a fulminant form of MS. Consistent with this observation is a report that PAD2 is significantly upregulated in MS brain. MBP has, by my count, twenty-nine arginine residues. If MBP is heavily citrullinated, its surface charge is changed dramatically, with potentially significant effects on myelin structure and function. Indeed, overexpression of PAD2 in oligodendrocytes is sufficient to induce demyelination in mouse brain.
 So our view would be that MS is indeed an inside-out disease and that activation of PAD2 is an early event in the injury cascade. Increased citrullination of MBP and other myelin components is sufficient to initiate a program of demyelination. In some patients, citrullination of MBP also results in the generation of neo-antigens, so that in these patients an autoimmune response is superimposed on the CNS-intrinsic demyelination program. It’s the autoimmune lesions and their clinical consequences that neurologists see and therefore what they treat. But if you buy this model, then treating patients with a PAD2 inhibitor could be a very effective strategy, certainly for progressive MS and likely for relapsing remitting disease as well.
So our view would be that MS is indeed an inside-out disease and that activation of PAD2 is an early event in the injury cascade. Increased citrullination of MBP and other myelin components is sufficient to initiate a program of demyelination. In some patients, citrullination of MBP also results in the generation of neo-antigens, so that in these patients an autoimmune response is superimposed on the CNS-intrinsic demyelination program. It’s the autoimmune lesions and their clinical consequences that neurologists see and therefore what they treat. But if you buy this model, then treating patients with a PAD2 inhibitor could be a very effective strategy, certainly for progressive MS and likely for relapsing remitting disease as well.
The Padlock seed germinates
That’s the science that got us all excited — and lured me back to my old flame, immunology. Atlas Venture had identified this as a project of interest in a discussion last year with the tech transfer team at The Scripps Research Institute. Two Scripps scientists were doing pivotal work on the PADs. Paul Thompson, who was at Scripps’s Florida site (he’s now Professor of Biochemistry and Molecular Pharmacology and Director of Chemical Biology at University of Massachusetts Medical School), is the world leader in the biochemistry of these enzymes — he knows these molecules literally inside and out. Kerri Mowen, Assistant Professor of Chemical Physiology and Immunology and Microbial Sciences at Scripps La Jolla, has done critical work on the biology of PADs. We incorporated the company in January, with Atlas, Scripps, Paul, Kerri, and Todd Huffman of Scripps Advance as founders. Atlas put in some seed money and we went to work.
The most immediate risk in front us was whether the PADs are even druggable; others had tried, we’d heard. If they’re not, there’s little point in forming a company to develop PAD inhibitors. Consequently, the focus of Padlock’s seed phase was to answer that question. We conducted full high-throughput screens against two different PADs at Evotec, developing all the necessary assays as well as conditions for structural biology. In parallel, we continued our deep dive into PAD biology and built relationships with key academics in the field. We gathered a group of these investigators in May for a PAD Summit — it was the first time many of these people were in the same room. And, of course, we lined up some money. We closed on our Series A financing back in April, bringing JJDC and MS Ventures into the syndicate along with Atlas, but we only pulled down enough cash to fund our seed activities. Lastly, we were fortunate to be able to assemble a first-rate management team, folks who were crazy enough to sign up at risk and agree to work only part-time (more accurately, to be paid only part-time) during the seed phase to help us conserve capital.
But now we are ready to fledge and fly. We’ve got plenty of exciting chemical matter, assays and reagents in place, a fantastic management team, a committed network of collaborators, and a savvy and deep-pocketed investor syndicate (just augmented by Index Ventures). We aim to break the science wide open and bring fundamentally new (I like to call them orthogonal) medicines to patients who badly need them.
Immunology, let’s make beautiful medicines together.
And now a word from our sponsor
Today’s press release can be found here. And our brand-spanking new website is here.