



This blog was written by Adam Rosenberg, CEO of Rodin Therapeutics and Atlas EIR, as part of the From The Trenches feature of LifeSciVC.
In my February 2016 post, I wrote “if some of the current generation of high-profile, expensive clinical trials fail to reach positive endpoints, we may have to work through some retrenching in this area.”
The clinical failure of Lilly’s solanezumab was a disappointment to all who hope for effective medicines in the fight against Alzheimer’s disease. As a result, some declared an end to the amyloid hypothesis, while others remain optimistic.
For this blog, I will not wade into the continuing, decades-long battle between the tau-ists and ba-ptists, or argue the nuanced distinctions between clinical trial designs or antibody properties, selectivity or brain penetrance. For a small selection of perspectives, see:
- Eli Lilly’s Alzheimer’s Antibody Does Not Work
- Failed Alzheimer’s Trial Does Not Kill Leading Theory of Disease
- What Does the Failure of Solanezumab Mean
Instead, I will attempt to bring perspective to the research, investment and translational impact of recent clinical results, primarily for those of us outside large pharma attempting to advance novel Alzheimer’s approaches towards the clinic.
In particular, I will argue that the field as a whole has overallocated resources to the amyloid hypothesis and that we need a more diversified, cost-effective approach, prioritizing therapeutic strategies that have the potential for meaningful and sustained patient benefit, without requiring quite so costly mega-trials.
Research and Investment Implications
As I previously noted, there are encouraging signs of a coming neuroscience renaissance. Recent announcements demonstrate both new company creation and funding, and continued pharma investment. As just a few examples, see:
- Abbvie’s New Foundational Neuroscience Center in Cambridge
- Eisai’s new Discovery Innovation Center in Andover
- Denali’s $130m Series B Round
On the Abeta front, Biogen and Roche continue development of their respective antibodies, and Lilly is pressing on with an alternative approach: (here).
Given differentiated precise targets and mechanisms, the continued investment in other clinical-stage, anti-amyloid antibodies should be applauded. But with each late-stage clinical failure of an Abeta-directed approach, it becomes clearer that we need more therapeutic diversity now, regardless of the fate of currently active programs.
Indeed, the nonprofit funding community has for some time diversified its approach beyond the amyloid hypothesis: (here). Academic translational centers are also pushing forward, notably Vanderbilt’s laudable effort to go it alone into the clinic: (here).
This is crucial, because while an amyloid-targeting approach may ultimately be approved, it is very unlikely to be a silver bullet. An approved antibody may be part of an arsenal or cocktail approach targeting different aspects of Alzheimer’s pathologies; but it should not be expected to “cure” Alzheimer’s disease in all patients at all stages of disease. Some patients with severe dementia have little Abeta pathology, and some patients with severe Abeta pathology have no dementia. And even with a tighter correlation of pathology and symptoms, because of the preventative nature of a proteinopathy-driven strategy, many patients will be too advanced at the point of diagnosis to expect a therapeutic benefit from amyloid intervention.
Thus, the medical need and commercial opportunity for continued, diversified Alzheimer’s research investment will remain as strong as ever, regardless of how the current amyloid-targeted mAb and BACE inhibitor trials read out.
Neurology is complicated, and requires a diversified investment and portfolio strategy
Translational Implications
We should learn from expensive Alzheimer’s clinical failures to craft more effective translational strategies going forward.
Ideally, programs now advancing into the clinic:
- address mechanisms/pathologies other than Abeta, that directly correlate with patient decline;
- have the potential to generate sustained patient benefit;
- are complementary to other marketed and clinical-stage therapeutics; and
- do not require mega-trials of 18-24 months to show a meaningful clinical effect.
I am squarely in the camp that believes the oversimplified “disease-modifying” vs. “symptomatic” distinction is obsolete. Yet such programs might fill a middle ground between temporarily addressing symptoms at one end of the spectrum, and curing or preventing disease at the other.
This perspective crystallized about 10 years ago, when I was with Link Medicine and we went to FDA to discuss an alpha-synuclein clearance strategy for potential disease-modification in Parkinson’s disease. We were squarely informed that reduction of synuclein (or amyloid, tau or huntingtin for that matter) is effectively meaningless from a regulator’s perspective without a corresponding clinically meaningful improvement for patients.
While regulatory perspectives have clearly evolved since then, it still seems unlikely that FDA will approve a CNS therapy based purely on biochemical biomarker endpoints, at least until the clinical relevance of that biomarker is conclusively proven.
This chicken-and-egg situation makes it highly challenging for even a well-funded venture-backed company – much less a company supported by NIH, foundation or angel capital – to show both proof of mechanism and clinically meaningful patient improvement in well-powered studies, especially with a proteinopathy-targeted approach.
Synaptic Resilience – A Case Study
Billions of dollars have been invested in amyloid antibodies, to the relative exclusion of alternative and complementary strategies
All of this supports the view that we need to learn from the amyloid clinical precedent, while focusing on our ultimate (and obvious) therapeutic goal, which is making patients better, or at least slowing their decline.
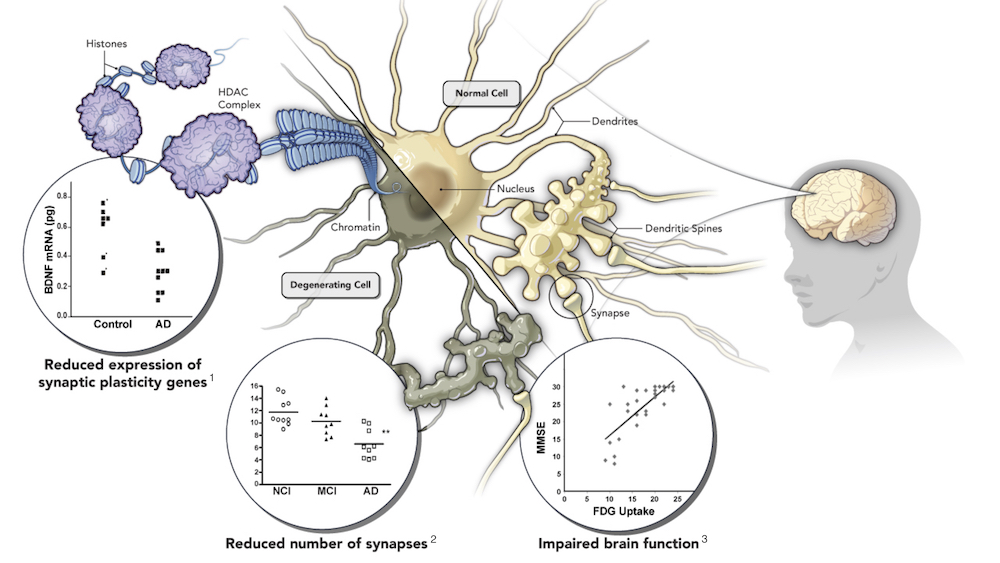
In Alzheimer’s disease, synaptic pathology is a major contributor to clinical symptoms such as cognitive decline, and is driven by molecular, structural and functional deficits
Might there be ways to directly improve synapse health, rather than just targeting alleged upstream adversaries?
At Rodin Therapeutics, we believe the answer is yes…with a therapeutic strategy directly targeting synaptic resilience:
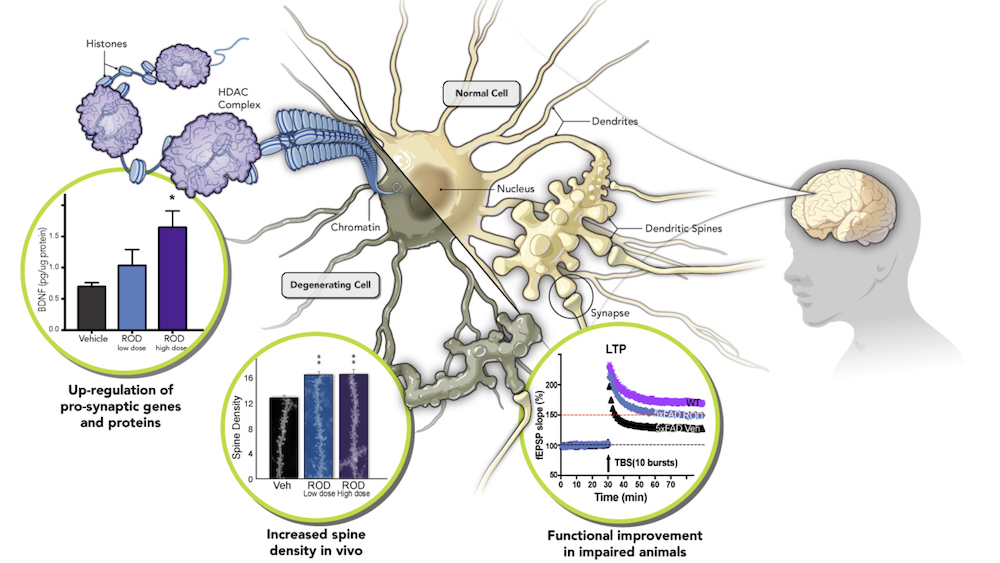
I’ll freely admit this might be slightly self-serving (but at least I’m voting with my feet!). At Rodin, we believe that directly increasing synaptic resilience (in our case, via epigenetic modulation) will produce sustained cognitive and/or functional improvement, without the need for continuous receptor/target occupancy (as is typically required for neurotransmitter-driven approaches). We are also directly addressing synaptic function, rather than trying to clear plaques or tangles with the expectation that protein disaggregation will ultimately lead to improved synaptic function.
We believe this strategy is therefore more amenable to an early efficacy go/no-go decision in the clinic, well before billions of dollars need to be invested for multi-year Phase 2/3 studies…but still with the potential to ultimately show arrest of cognitive and functional deterioration. And we believe that our compounds also have potential to treat many diseases beyond Alzheimer’s, where synaptic density and dysfunction (as a convergent result of different pathological mechanisms) are a proximate cause of neurological impairment and disease progression.
Rodin’s approach to synaptic resilience is just one example where sustained benefit may be demonstrable with reasonable clinical development costs and timelines. This is also well worth tackling with approaches targeting neuroinflammation, vascular disease, and mitochondrial dysfunction, for example.
Clinical Implications – An Ambitious Wish List
While nothing is easy in neurodegeneration R&D, I have a relatively succinct request for the next generation of Alzheimer’s clinical development: we need many more carefully designed, clinic-ready, brain penetrant small molecules, with good PK and chemical properties and a reasonable therapeutic index, to go beyond the amyloid hypothesis and test more diversified approaches.
An ambitious wish list would also include therapies that can work across a heterogeneous patient population, in all disease stages, with efficacy both alone and as a complement to drugs targeting adjacent mechanisms.
Broad addressable patient populations:
In 50 years, Alzheimer’s disease (as we currently, rather monolithically, conceive of it) will probably be re-defined as a series of more specific disorders, more precisely described in terms of genetics, pathology, and clinical symptomology. Until then, it is highly advantageous to advance therapies with potential efficacy across heterogeneous patient populations, regardless of ApoE4 status, tau and/or amyloid pathology, etc.
Efficacy across disease stages, including in later-stage patients
Clinicians and drug developers have largely concluded that later-stage patients may be too far advanced to benefit from therapies targeting upstream proteinopathy-related pathology. So the need remains high for therapies providing lasting cognitive and functional improvement for patients with active disease. Strategies focused on synaptic resilience, neuroinflammation or mitochondrial function, for example, may target Alzheimer’s cognitive defects more directly, and therefore be less reliant on intervening very early in disease progression.
Complementary to other therapeutic approaches
Finally, while new Alzheimer’s therapies will likely need to show meaningful benefit as a monotherapy, the ultimate goal in treating this heterogeneous disease will likely require a cocktail approach. Ideally, new clinical candidates should therefore be complementary with other therapies targeting adjacent mechanisms, like neurotransmitter modulation or clearance of toxic protein aggregates.
Conclusions
These are ambitious goals, to be sure. But now is the time to take stock of what we’ve learned from the late-stage trials of amyloid-targeting drugs, and apply those learnings to the development of more diverse therapeutic approaches. We must recognize that there has been a historical overallocation of resources towards the amyloid hypothesis. We should accelerate development of alternative strategies that may yield benefit in broader patient populations, with shorter/smaller trials, and which can ultimately complement proteinopathy-directed therapies if they are eventually proven efficacious.
Figure data references:
1. Phillips HS, Hains JM, Armanini M, Laramee GR, Johnson SA, Winslow JW. BDNF mRNA is decreased in the hippocampus of individuals with Alzheimer’s disease. Neuron. 1991 Nov
2. Scheff SW, Price DA, Schmitt FA, Mufson EJ. Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol Aging. 2006 Oct
3. Newberg AB, Arnold SE, Wintering N, Rovner BW, Alavi A. Initial Clinical Comparison of 18F-Florbetapir and 18F-FDG PET in Patients with Alzheimer Disease and Controls. J Nucl Med. 2012 Jun