



The FDA recently approved Imfinzi (durvalumab) for the treatment of bladder cancer, making it the fifth new monoclonal antibody approved this year. More than twenty antibodies have been approved by the FDA since 2015. While not quite the magic bullet envisioned by Ehrlich over a hundred years ago, antibodies have become a critically important modality for high impact medicines. With AstraZeneca’s durvalumab success, it’s worth reflecting on the state of antibody discovery and development in the industry today.
For those tracking R&D productivity metrics, antibodies have been a real outperformer: based on a comprehensive data review of clinical success rates, antibody (mAb) programs were found to have a likelihood of approval at IND filing 2x higher than conventional small molecule NCEs (14.1% versus 7.6%, respectively). Further, non-oncology mAbs were even more likely to succeed, exhibiting a 19.3% probability of going from IND to market.
While these metrics are great, and reflect some of their intrinsic, natural advantages as therapeutic agents, there’s still considerable rates of failure: 80-85% of mAbs are discontinued during development. Recent failures of Oncomed’s demcizumab (and earlier tarextumab and vantictumab) and Astrazeneca’s tralokinumab Phase III asthma trial are just the latest examples, and highlight the challenge of drug R&D with any modality. And even once they are approved, failures can continue happen – like Roche’s unexpected failure with Tecentriq in bladder cancer this week.
To frame the mAb landscape, a quick background on the various mAb frameworks is warranted. Early mAb therapeutics were derived from mouse immunizations, which led to the first approved agent, muromonab-CD3 (OKT3). As “foreign” to humans, these murine antibodies were highly immunogenic, leading to anti-drug antibodies (called human anti-mouse antibodies, or HAMA) that neutralize the mAb and prevent it from working effectively. The quest to reduce a mAb’s immunogenicity by relying on increasing amounts of human antibody sequences became an important vector in the field’s evolution since the 1980s. Chimeric antibodies, which use mouse variable domains and human constant regions, were a step forward, but these too are typically immunogenic.
{kind=link}

Sketches of chimeric (top right), humanized (bottom left) and fully human (bottom right) monoclonal antibodies. Human parts are shown in brown, non-human parts in blue. Source: Wiki Commons
Humanization techniques help convert mouse-derived antibodies into less immunogenic constructs, largely by limiting the murine sequences to just the CDRs (the binding loops) in the variable domains. It’s well established that humanized mAbs have lower immunogenicity than chimeric or murine formats. Fully human mAbs, where the entire sequence derives from human genetic repertoire, were developed originally via transgenic mice or phage display, and are believed by many to be the “preferred” mAb format. However, to my knowledge there’s no definitive data demonstrating that fully human mAbs are less risky for immunogenicity than humanized constructs – though the perception exists, especially as a rhetorical differentiation message versus mAbs of other formats.
Today there are at least 56 FDA approved antibodies by my count. Humanized mAbs represent 44% of these (25 products). About half of these came out of humanization technology from PDL BioPharma, which was broadly licensed. Genentech’s antibody portfolio has largely derived from humanized mAbs using PDL’s approach: Avastin, Herceptin, Lucentis, Xolair, Kadcyla, and Perjeta. UCB, from its Celltech roots, also has an incredibly strong antibody platform (derived from SLAM) leveraging immunization and humanization techniques as well as deep high throughput sampling of B-cell mAb repertoires.
While humanized antibodies remain important (and will for the foreseeable future), fully human constructs are increasingly common, and growing as a proportion of mAbs in the clinic. Since the approval of the first fully human mAb in 2002 (adalimumab/Humira), 23 fully human mAbs have been approved, or 40% of all of the marketed mAbs today (note: updated from original post from 22 to 23). Four of the five antibodies approved this year were fully human in origin, and 11 of the 21 mAbs approved since 2015.
As mentioned above, fully human antibodies have historically come from two discovery methods: immunization of transgenic mice which have been engineered to express human VDJ antibody sequences, or via screening of antibody libraries derived from human B-cell repertoire and presented via phage display. These two platforms have their advantages and disadvantages. Transgenics benefit from all the in vivo forces of selection (like diversity, expression, and stability), but antigenicity (generating an immune response) can be an issue. Further, transgenics are more difficult if positive counter-screening (like species cross-reactivity) is important. For phage, its vice versa in most cases. And both have been productive on similar targets. As an example, AZ’s durvalumab was discovered with the Xenomouse platform from Abgenix after immunizing for human PD-L1. Pfizer and Merck Serono’s recently approved avelumab, also against PD-L1, was discovered via Dyax’s phage display platform.
To probe the relative contributions of these different fully human platforms, the origins of the 23 fully human approved antibodies were explored, captured in the chart below. The striking observation is that transgenic immunizations have delivered nearly three-quarters of new fully human mAb approvals versus phage display.
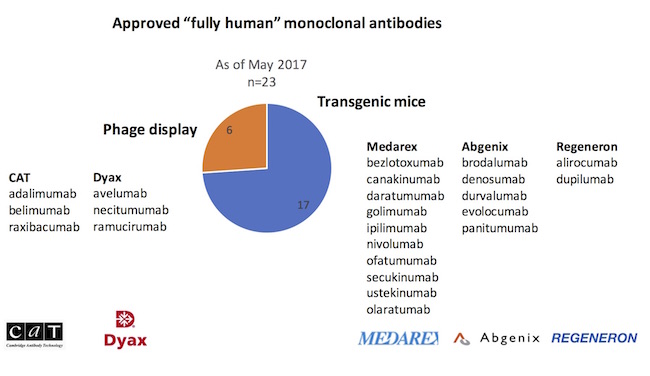
In particular, it’s worth highlighting the transgenic UltiMab technology developed at Medarex. With ten product approvals*, Medarex’ UltiMab platform has produced more approved drugs than any other human antibody platform in the industry. As an aside, BMS’s acquisition of Medarex for $2.6B in 2009, viewed with some skepticism at the time, is probably one of the best biotech acquisitions of all time. Nivolumab (Opdivo), discovered out of the Medarex platform, had sales of $2.6B in the US alone last year, and is forecast to have sales north of $12B within five years. Funny to reflect on this wonderful lead sentence about Medarex in an article in the spring of 2009: “If there’s a pharma company that rewards executives for delivering absolutely nothing, thy name is Medarex.” Delivering nothing? How about one of the most productive antibody platforms ever created.
An intriguing question, though, is why have transgenic platforms outperformed phage display in the number of new product approvals by such a large margin.
It’s not the differential gestation period of the technologies. Both are two decades old at the very least, as all of the six major players were founded over 20 years ago: Abgenix (1996), Medarex (1987), and Regeneron (1988) in the transgenics area, with Dyax (1989), CAT (1989), and Morphosys (1992, an Atlas Fund II investment) in the phage display space. Both technology approaches suffered through painful patent battles between the players. And all had their first wave of early mAbs enter development more than 15 years ago. So the technologies are similarly mature in many ways.
It’s also not because the two platforms had massively divergent BD strategies, at least at the macro level. The transgenic players were aggressive deal-makers around multi-target collaborations (e.g., Abgenix-AZ’s 2003 deal around 36 targets, Medarex-Pfizer’s 2004 deal around 50 targets, Medarex-Centocor/J&J’s 1997 deal that was extended in 2007 to an unlimited number of targets, etc…). But so were the phage players, who did lots of big multi-target deals too (e.g., CAT-AZ’s 2004 deal preceding their 2006 acquisition, Morphosys-J&J/Centocor’s 30 target deal in 2000/2005, Dyax with Biogen/Cubist/others). It’s interesting to speculate about whether subtly different deal structures across these players altered the future success of these platforms at bringing programs into clinical development (e.g., different target exclusivity provisions or target-swapping mechanisms, different resourcing obligations or R&D diligence requirements, different tech transfer rights to the platforms in their own labs); however, although there might be something in the detail, I’m doubtful deal-making differences contribute significantly to the large delta in approvals though.
But it might be the characteristics of the antibodies themselves. Anecdotally, phage-derived antibodies suffer from a higher frequency of biophysical property issues that create barriers to “developability” of antibodies. These suboptimal attributes can lead to difficulties in PK (like accelerated clearance limiting time “on target”), safety, CMC, and manufacturing issues. But data on these anecdotes is rather scant.
A fantastic 2017 paper from Dane Wittrup and colleagues at Adimab (here) characterized a large set of clinical-stage antibodies for their biophysical properties. In an attempt to identify some guidance criteria for developability of mAbs, much like Lipinski’s Rule of Five did for small molecule physiochemical qualities, the authors ran each antibody through a panel of assays, which I’ll call the Dirty Dozen. Simplifying a great deal, these Dirty Dozen assays measured the promiscuous stickiness of antibodies (both self-interactions and “poly-specific” cross-protein interactions), the greasiness of the antibodies (hydrophobicity), protein complex formation (monomer vs aggregates), thermal stability (melting temperature), and recombinant expression levels. Across most of the Dirty Dozen, the distribution of data wasn’t Gaussian, but instead was skewed with long low-frequency tails out into “unfavorable” ranges. Using marketed antibodies to create a standard curve, any antibody with a Dirty Dozen parameter in the worst 10% of the range was considered as possessing a “red flag” – and they profiled how many red flags different antibodies had. As shown on the left below, approved antibody drugs had fewer flags (nearly 60% had no flags on any assay, less than 10% had 3 or more) – while Phase 2 stage antibodies had a much higher frequency of flags. This type of trend where earlier stage investigational drugs have a wider range of unfavorable characteristics is common, and the intuitive expectation is that those traits lead to higher development risk profiles over time. Interestingly, the authors also compare in vivo mammalian-derived mAbs to phage display and the same profile emerges: phage-derived mAbs have more biophysical red flags than those from in vivo sources like transgenic mice (right side below).

However, at least to date, these unfavorable biophysical traits haven’t led to a pronounced statistical difference in clinical attrition or success rates, although the existing datasets are small and largely unpublished. Evaluating the current status of the 64 fully human antibodies that were in clinical development in 2008, as profiled by Nils Lonberg in this seminal review paper (so nearly 9 years later), we found that the relative success rates were almost identical across transgenic and phage-derived antibodies: roughly half were discontinued, a quarter still active in development, and a quarter approved. Only time will tell if the anecdotes and data around biophysical properties plays through.
So what’s the future of antibody discovery?
Like many drug discovery fields, there’s been a continued quest to improve the quality, diversity, and developability of antibodies derived from these fully human methods.
Early generation transgenic mice, while very productive, lack native B-cell signaling (i.e., human Fc regions of their mAbs do not signal as effectively through the murine B-cell receptors) and therefore are often “hypo-responders” to certain antigens/immunizations. They also didn’t carry the full human VDJ repertoire. Companies like Harbour Antibodies (an Atlas Fund IX investment), Kymab, Ablexis, and more recently Trianni are all working on next generation transgenic murine models, and OMT has done same with rat transgenics, that attempt to address these issues.
Early phage libraries suffered from sequence diversity issues, the biophysical liabilities mentioned above, and a lack of native pairing of heavy and light chains. Many players out there are advancing solutions to these issues, including both existing phage players like Morphosys as well as startups like Distributed Bio.
To my knowledge, none of these “new” transgenic or phage display companies have their fully human antibodies in the clinic yet – but over the next twelve months we expect to see a number of first-in-man studies with antibodies from these new platforms.
Lastly, over the past eight years other platforms have emerged as well. Of note, the yeast display techniques at Adimab have proven themselves to be highly productive – with more than a dozen antibodies active in clinical development today. They are one of the most prolific antibody providers in the business today, and widely viewed as one of the best.
As many of the firms in the original wave of fully human antibody providers have been acquired (CAT, Abgenix, Medarex, Dyax), the risk of not having access to these platforms became a real concern for new mAb-product focused companies, especially startups. Fortunately, the next generation of antibody providers (some listed above), and the proliferation of fully-enabled antibody discovery contract partners, has provided the product-focused biotech sector with plenty of choices. And interestingly, a number of CROs are starting to offer some of the assays in Wittrup’s Dirty Dozen as part of their standard characterization battery.
Most antibody discovery efforts today parallel track a number of methods: conventional DNA-immunization, transgenic rodents, phage display, and engineered yeast-derived approaches. Across our Atlas portfolio alone, we have deals with Harbour, Trianni, Adimab, ChemPartner, Wuxi, Genscript, Aldevron, Lake Pharma, and many others.
In summary, after a few decades of R&D, therapeutic human antibody discovery techniques have fully come of age, with a broad range of productive platform offerings. Nearly two dozen fully human mAbs are approved for use in a wide range of diseases today, and scores more are in development.
Instead of worrying about “can I make a potent antibody”, the focus for most researchers has shifted to “will a potent antibody deliver value to patients”. This latter question is where most antibodies, if they fail, come a part in the clinic these days: disease biology is hard to fully appreciate until you do the clinical work. Even against the same antigens/pathways, the interaction of antibodies with biology is complicated: as exhibit A, take Tecentriq’s and Opdivo’s recent failures in settings where Keytruda has worked. Once again, like with computer-aided small molecule drug discovery, biology and its incredible complexity consumes all – even “magic bullet” antibodies.
*Note: Number of fully human antibodies approved was updated after the original post from 22 to 23 mAbs, with Medarex’ platform contributing 10 (rather then 9) products. Thanks to Nils Lonberg for the correction!