



This blog was written by Jonathan Montagu, CEO of Hotspot Therapeutics and Atlas EIR, as part of the From The Trenches feature of LifeSciVC.
It starts off with flu-like symptoms and progresses to a perpetual low-grade fever, excessive sweating and loss of appetite. A physical exam reveals a grossly enlarged spleen while basic blood work uncovers white blood cell counts between thirty and sixty times normal levels.
Historically, patients with CML would live only three to five years, during which time the disease would quickly transform from a chronic leukemia to an aggressive and fatal acute leukemia. Thankfully, CML became the poster child for targeted cancer therapy, as molecules designed to specifically target the ATP-binding active site of Abl1 led to approval of imatinib Gleevec® in 2001. CML patients now enjoy roughly 84% ten-year survival. With peak sales of $4.6B, Gleevec was so successful that it created a new market.
However, despite the incredible advances in CML treatment, most patients remain destined to stay on potent kinase inhibitors for the rest of their lives. Furthermore, T315I and other resistance mutations occur in a subset of patients for which only the black-box labelled ponatinib Iclusig® is currently approved. Despite the incredible benefit of these targeted therapies, we have stopped just short of a cure.
Until now.
In March of last year, Andrew Wylie, Joseph Schoepfer and colleagues at Novartis reported data from asciminib (ABL-001), a radically new approach for targeting oncogenic fusions of Abl1. Asciminib engages the kinase in a way that is entirely distinct from the enzyme’s active site. As such, asciminib is an allosteric inhibitor and its mechanism of action mimics the way that nature itself controls the protein through the addition of a fatty tail (myristic acid).
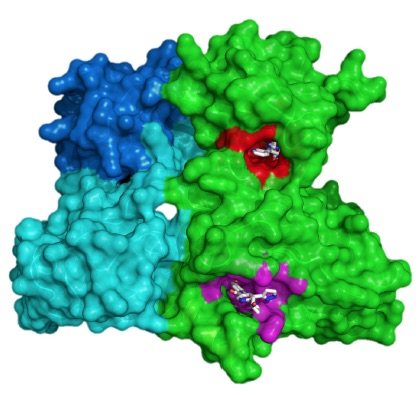
X-ray crystal structure of ABL001 bound to the myristoyl pocket in magenta and nilotinib bound to the ATP pocket in red co-crystallized with ABL1. Source: Wylie et al. doi:10.1038/nature21702
Given its unique mode of action, asciminib has a distinct resistance profile when compared with traditional therapies and achieves low nanomolar activity against tumours harbouring the T315I resistance mutation. In early clinical studies, asciminib has driven robust anti-tumor responses as a single agent in heavily pre-treated patients, including those resistant or intolerant to existing kinase inhibitors.
However, things get really interesting when one combines the allosteric inhibitor asciminib and the active site inhibitor, nilotinib. Due to the complimentary resistance profiles, this combination led to complete disease control that eradicated CML xenograft tumors without recurrence after the end of treatment. For those of us who are used to looking at xenograft data, it is hard to imagine a more compelling picture.
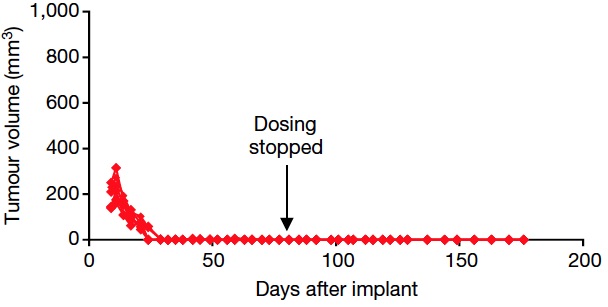
Treatment of KCL-22 mouse xenografts with asciminib 30 mg/kg and nilotinib 75 mg/kg. Source: Wylie et al. doi:10.1038/nature21702
Assuming these observations translate in the clinic, the significant implication of these data is that combination therapy with asciminib could result in clinical cures.
This is not the first time that a breakthrough combination regimen has been uncovered in preclinical models. Rather, the challenge has been finding a combination regimen that is tolerable because the adverse-events of drugs can ‘stack’ on top of each other, as was discovered with the first anti-CTLA4 and BRAF combination study in melanoma. In the case of asciminib, there is good reason to be optimistic of clinical success. The pocket that asciminib binds to appears unique – allosteric sites are far less conserved across evolution than active sites – resulting in an agent with an exceptionally clean selectivity profile. Furthermore, by avoiding the enzyme’s active site, we are liberated from the challenging chemical space that has plagued drug discovery for decades.
The challenges of allosteric drug discovery
Allosteric targeting of BCR-ABL has resulted in the potential for an exceptionally well tolerated, safe medicine that could deliver clinical cures for the first time in the history of CML. So, given all the inherent benefits of allostery, why aren’t we all abandoning active site-targeted drug discovery? There are a number of underlying reasons for this.
The term allostery comes from the Greek allos, meaning “other” and stereos, meaning “solid object”. This refers to the fact that allosteric modulators have their effect outside the active site. But the question is where exactly? Therein lies the challenge. Active sites are easy to discern because we have learnt what to look for. Allosteric sites remain largely hidden, at least to the untrained eye. Sometimes these pockets are present in x-ray crystal structures, but we lack the understanding of the functional role of these regions.
Much of the established drug discovery paradigm is geared towards the identification and progression of active-site inhibitors. Corporate chemical collections are largely overpopulated with active-site directed scaffolds from decades of efforts against these sites. The deck is essentially stacked against the discovery of allosteric modulators from the start.
High-throughput screening approaches used to screen such libraries have focused on assay readouts that can readily scale to millions of compounds. In this context, all efforts are made to optimize the signal-to-noise ratio. This means reducing what we study to simpler, well-behaved systems. Eliminating ‘troublesome’ regulatory domains from a full-length protein often makes these assays more scalable, but the protein being studied starts to look less and less like the natural protein.
Significant organizational and cultural barriers work against the development of allosteric inhibitors. At the outset, designing a screening paradigm to uncover allosteric modulators requires work up front to understand the structure-function of the protein at hand and establish counter screens to select molecules with the desired mode of action. In the era of metrics and productivity, and competing demands for internal screening capacity, programs that do not deliver immediate satisfaction are less likely to progress.
Active site inhibitors will frequently exhibit good potency out of a screen. Medicinal chemists understandably focus their attention on these ‘gifted’ molecules despite the fact that their time in the limelight can be short lived due to inherent selectivity challenges and poor cellular potency. Allosteric inhibitors require more care and nurturing. At the outset, they are often less potent, take more effort to achieve nanomolar potency, and may well break the sacred Lipinsky rules. For these reasons, few discovery groups can avoid the gravitational pull of the active site.
New directions for allostery
So what can be done to address these challenges? There are reasons to be excited about the future of allosteric drug discovery.
Seeing what’s in plain sight: New approaches are being evaluated to identify allosteric pockets. For example, insights from how nature itself regulates proteins point to attractive allosteric sites. It is notable that asciminib interacts with precisely the same site that nature uses to regulate the enzyme. By targeting these ‘regulatory hotspots’, one has confidence that the site on the protein really matters. Similarly, the Acetyl CoA Carboxylase (ACC) program led by Gerry Harriman while at Nimbus Therapeutics, targeted a regulatory site used by nature to control the enzyme. This strategy led to highly selective and potent allosteric molecules, the most advanced of which is currently in Phase 2 for the treatment of NASH.
Relay Therapeutics is coming at the problem from first principles. Using specialized hardware and software, Relay simulates protein movement over long periods of time (seconds) which can uncover cryptic pockets not visible in static x-ray crystal structures.
Traditional fragment-based screening can be used to sample protein conformations experimentally which, while time consuming and expensive, has uncovered allosteric pockets. Disulfide trapping, pioneered by James Wells and the team at Sunesis, allows specific parts of a protein to be probed for pockets. Although there is nothing new about these two approaches, we are starting to see the fruits of these endeavors. A couple of weeks ago, the Wellspring, J&J and Shokat teams reported in Cell on the latest iteration of inhibitors targeting a mutated cysteine in an allosteric pocket of KRas.
A natural setting: If we can represent our biological targets in a way that better recapitulates their natural setting, we can give our chemistry more opportunity to shine. When Novartis first screened Abl1, the team led by Markus Warmuth and Nathanael Gray argued that screening the kinase domain in the same way that they and twenty other companies had done in the past, was unlikely to uncover anything new. Instead, they used a cell-based assay to identify GNF2, one of the precursors of ABL-001. This screening approach was critical since it allowed BCR-ABL to be presented as a full-length protein in its natural environment.
More recently, Phoremost has used cell-based screening of lentiviral libraries carrying millions of peptides. Phenotypes of interest turn the cells green via expression of green fluorescent protein (GFP) and the corresponding peptide is uncovered by sequencing the DNA. This feels like a DNA-encoded library done in cells which nicely solves the problem of displaying the target protein in the right cellular context.
Chemical diversity: It is important that chemistry libraries are well aligned with the requirements of allosteric pockets while avoiding typical active site scaffolds. The original GNF2 series came from a library of 50k compounds, while the most recent published SHP2 allosteric inhibitors were obtained through a screen of 100k diverse molecules, highlighting the fact that the right library is more important than the biggest.
Screening paradigm: Successful allosteric drug discovery requires elegant screening paradigms to rapidly identify compounds with the desired mode of action. Rather than simply asking whether the compound inhibits the target, a clever set of calibrated questions allow us to understand how and where. This is nicely illustrated by the NMR assay used by the Novartis team to assess the conformation of the critical C-terminal helix of Abl1.
To conclude, pursuing allosteric drug discovery is not for the faint of heart because it requires strong, committed research leadership that is comfortable breaking the conventional rules of drug discovery and can drive a bespoke and tailored scientific approach. The rewards are certainly there for the teams that take on this challenge. Hats off to the Novartis team for believing in ABL-001 and the remarkable potential that it holds in combination with the company’s Gleevec franchise. In the past, CML was a death sentence but allosteric inhibitors such as ABL-001 now hold the promise of a clinical cure.