



This blog was written by Jonathan Montagu, CEO and founder of HotSpot Therapeutics, as part of the From The Trenches feature of LifeSciVC.
The predictions around the impact of artificial intelligence (AI) have been broad reaching but it’s clear that not everyone is convinced (here and here) and the trajectory of the current AI hype cycle feels downwards.
In this blog, I want to make the case that artificial intelligence (AI) will make a significant difference in drug discovery over the long run but it’s important to see AI like other big industry shifts such as the genomics revolution of the early 2000’s. Incremental changes over time lead to big and even transformational impacts. Today there are structural constraints in descriptors, data and algorithms that are limiting the role of AI but in the longer term innovation in these areas will enable real impact. Overall, I am optimistic because I see what artificial intelligence has brought to other fields such as image analysis and speech recognition.
Déjà vu
As you read some of the AI headlines, it feels a lot like the early 2000s. The human genome had just been sequenced. We were swimming in new data. There was a sense of “more is more”. More sequencing data, more combinatorial library compounds, more zebrafish knock outs. We were convinced that surely something good would come from it all. And, of course, it did but it took a lot longer than we all thought. As reality set in, it was clear that owning a small piece of the value chain, e.g. a cloned or sequenced target, far removed from the product, wasn’t going to command much value. The big data companies like Celera came and went and were ultimately replaced with product focused companies such as Millennium (following the LeukoSite acquisition), Plexxicon, Exelixis and Array that took advantage of the genomics revolution but translated these insights into medicines.
AI and machine learning have a similar feel today. Lots of data, lots of promise … but we seem a long way from product value or clarity around which business models are going to succeed.
To make a big topic digestible, I’m going to focus on the incremental but important applications of AI in chemistry but many of the same themes apply to other applications of machine learning including target validation, drug design and clinical studies.
What is Artificial Intelligence?
At the outset, it’s important to be clear what we are talking about. The terms artificial intelligence (AI) and machine learning seem to be used interchangeably but there’s an important difference as shown in the figure below:
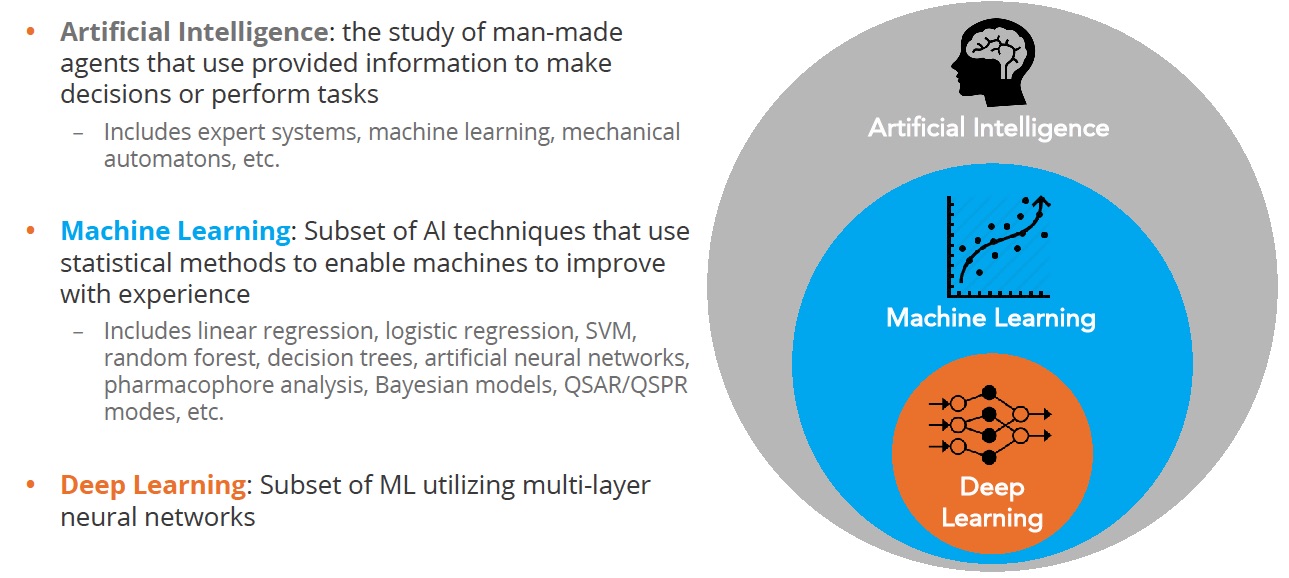 Figure 1 Deep learning is a subset of machine learning
Figure 1 Deep learning is a subset of machine learning
Artificial Intelligence comprises a large field aimed at recreating human intelligence in machines. Researchers take advantage of many flavors of algorithm including expert systems, machine learning, and others. The goal is to create systems that can interpret external information, learn from it and then adapt to achieve certain goals.
The dominant AI approach applied to chemistry problems is machine learning which, at its core, is a statistical method that looks for correlations between something that can be changed e.g. the structure of a molecule, and properties that are measured e.g. potency
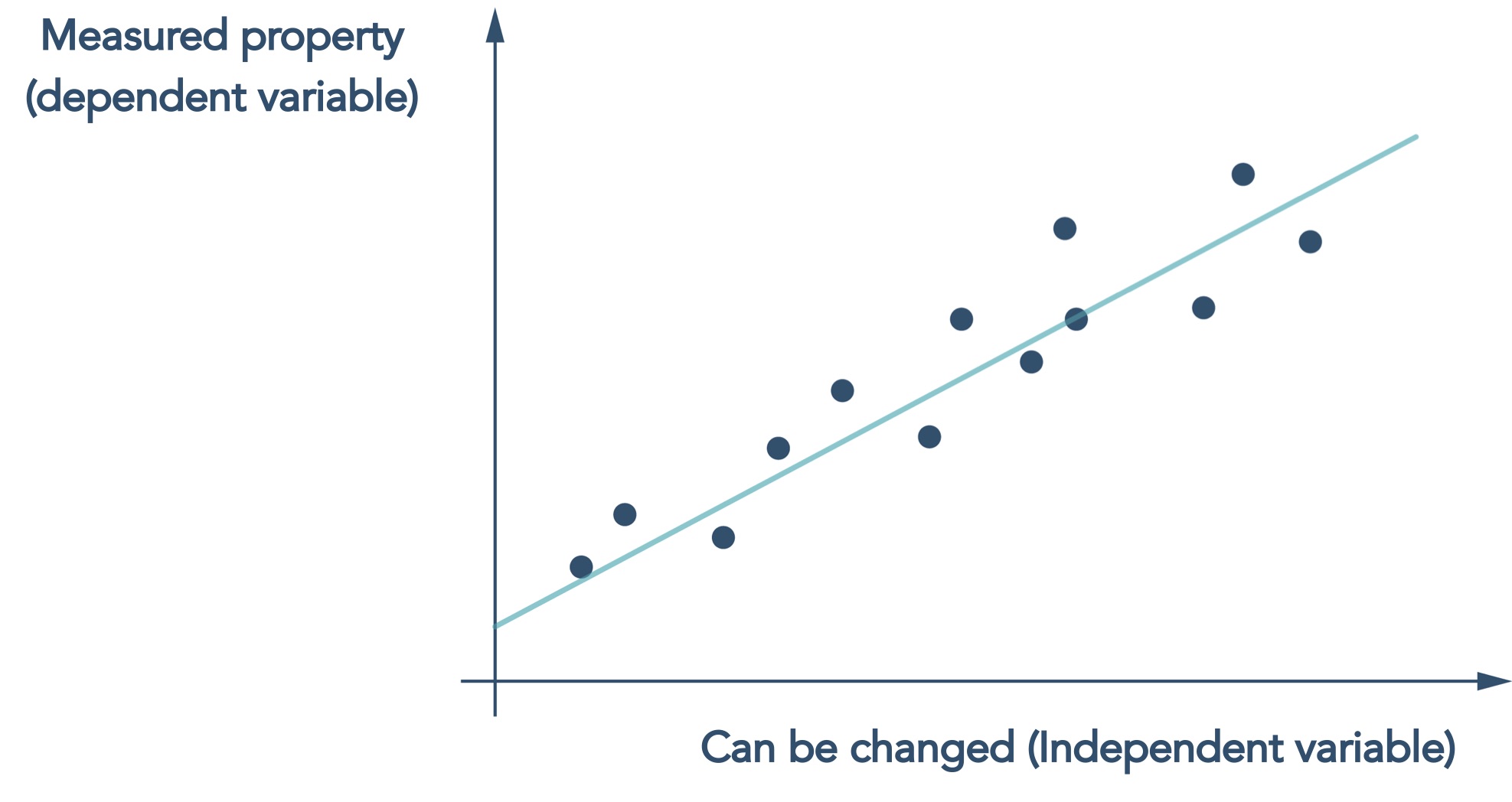
Figure 2 Machine learning models are essentially statistical regression models
Deep learning is a subset of machine learning that is loosely inspired by how the brain works through neural networks. Deep learning can tease apart features that are non-linear and correspond to different levels of abstraction. This is highly effective in image analysis where you can represent the features of, for example, a cat as point in multi dimensional space, and then predict the likelihood that what you are looking at is indeed a cat.
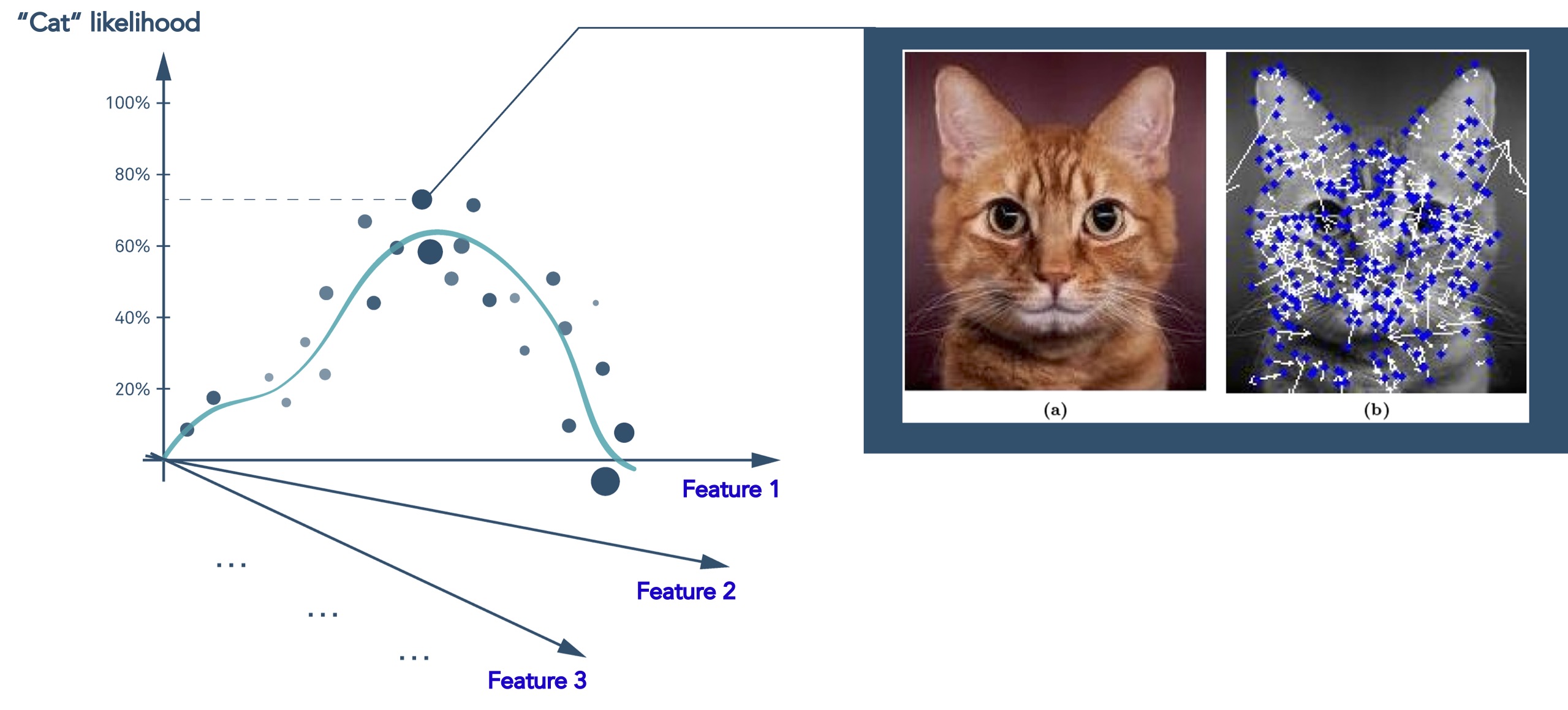 Figure 3 Deep learning models predict the likelihood that an image represents a cat using the same type of statistical regression analysis
Figure 3 Deep learning models predict the likelihood that an image represents a cat using the same type of statistical regression analysis
This may sound impressive but it’s important to remember that even the most complex neural networks are still regression models at heart. You are limited to what you train on. If you provide the above algorithm with a million cat images, it’s not going to be useful when presented with an elephant.
Applied to drug discovery, machine learning may uncover hidden patterns in existing data but it’s not going to offer creative leaps that go beyond the training data. “Machine learning can be powerful, but it’s not magic,” commented Ramy Farid, President & CEO of Schrödinger, Inc, a leader in computational drug discovery.
It seems unlikely that machine learning is ever going to emulate true intelligence; it might, but that’s not really the point. Things that humans do such as image processing, driving cars, reading, playing games such as chess and Go obviously require ‘true intelligence’. Attempting to emulate that type of intelligence with deep learning might actually work some day. On the other hand, predicting the affinity or solubility or clearance of a small molecule has nothing to do with human intelligence.
“The truth is that humans are not very good at predicting chemical and related biological properties such as affinity, solubility, and other such metrics because they are far too complex: no human can ‘see’ entropy, predict the dynamics of a protein, identify the locations of unstable waters in a protein binding site, and so on,” added Ramy Farid. “Physics-based methods that incorporate the principles of quantum mechanics and molecular dynamics are needed to accurately model and predict these complex molecular phenomena. In this context machine learning models can be quite powerful.”
This is where machine learning models can really shine, and we already have a great precedent: “We’ve effectively been doing machine learning in medicinal chemistry for at least forty years. It’s called QSAR”, said Jon Mason, Senior Research Fellow at Sosei Heptares, Ltd. Quantitative structure-activity relationships (QSAR) are how chemists correlate the structure of a molecule with its activity in an assay of interest, for example potency against an enzyme.
So what’s new and why all the fuss? “We now have protein-molecule X-ray structures for a large number of biologically active molecules,” Jon Mason noted. “This allows the use of AI descriptors that are far more relevant, and potentially causative, because they are based on properties of the protein-molecule complex, not just the molecule.”
Opportunities and constraints
As we look forward, there are essentially three technological factors that are driving and limiting the utility of machine learning in chemistry and other fields:
- Descriptors: how well you can describe the features of your molecule and the protein that it’s interacting with
- Data: the amount of relevant, real life data you have to train your machine learning system on
- Algorithms: the underlying methodologies for doing the machine learning and for generating descriptors
Descriptors
If you can generate rich descriptions of your molecules and target protein, you are more likely to uncover more insightful patterns in the data. This is illustrated by the fact that machine learning has been especially effective in image recognition, but it has taken decades to develop algorithms that can extract the right type of information from images. The state of the art is now an approach called the Convolutional Neural Network (CNN), a deep learning method that loosely resembles the organization of the animal visual cortex, where images are interpreted in different layers. The first layer distinguishes basic attributes like lines and curves. At higher levels, the brain recognizes that a combination of edges and colors is, for instance, a train or a cat.
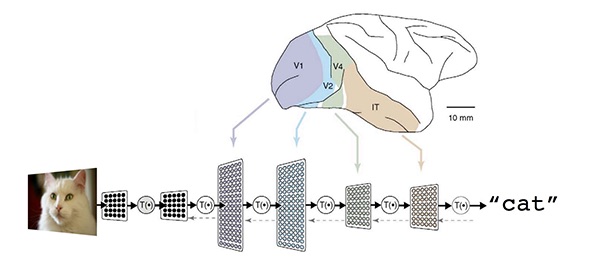
Figure 4 A convolutional neural network, source: Google Cloud
How does this translate to molecules and biological targets? If we are restricted to the flat, 2D world of molecules drawn on a piece of paper, our models are going to be pretty uninformative. For example, if we were to train a machine learning model on the series of molecules below, we would likely predict that Molecule A would have a potency in the 300 – 600 nanomolar range.
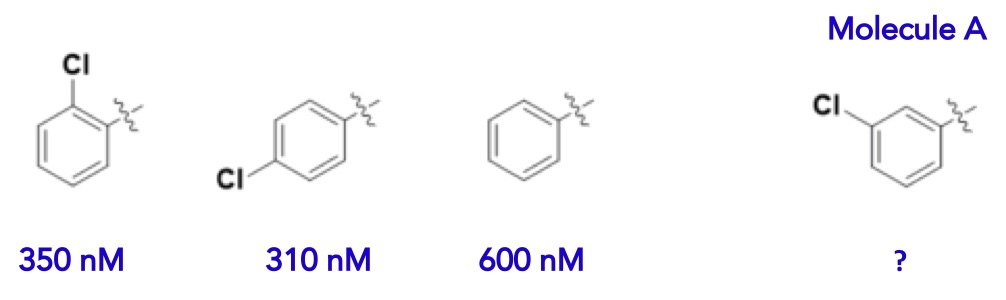
Figure 5 What’s the potency of molecule A?
However, when measured, this molecule is dramatically more potent (10 nM). Why is the model so badly wrong? The reason is that the model is lacking critically important descriptors of what’s going on in nature. The molecules above are literally living in a flat world.
Now imagine what happens if we consider that the world is not flat but three dimensional. Our molecules not only have 3D structure but they also interact with a protein that contains water.
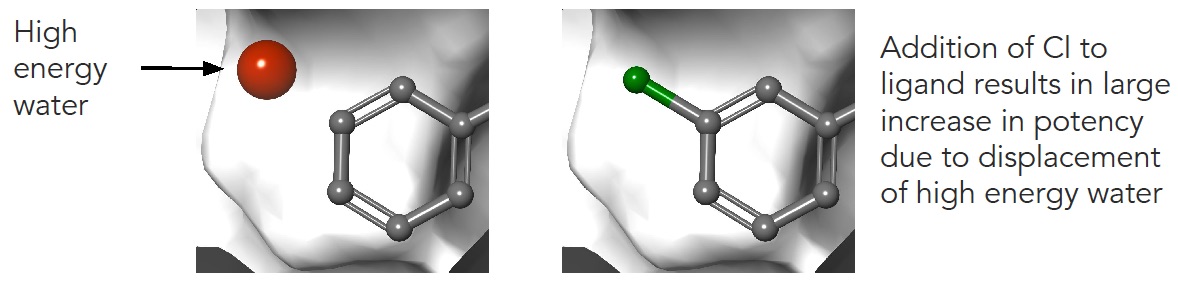
Figure 6 High energy or ‘unhappy’ waters are critically important in molecule binding and must be considered as descriptors in a machine learning model
We now immediately appreciate that Molecule A can kick out an “unhappy water” when it binds. If the energy released by this water were somehow provided as a descriptor, our machine learning model would likely get much closer to the experimental value.
In comparison to image recognition, the descriptors used in chemical machine learning models are still rather rudimentary. Much of the work to date has been done on text descriptions (1D) of molecules or flat 2D images. There’s also a fundamental difference between predicting affinity and recognizing an image. In the case image recognition, the cat or the dog is completely determined by the image itself. However, when presented with the 3D coordinates of a protein-molecule complex, information essential to the binding affinity is fundamentally not contained within that set of coordinates, for example the contributions of the red “unhappy” water above. Likewise, movement of the protein and molecule will generally make large contributions to binding affinity (through a thermodynamic concept called entropy) which cannot easily inferred from the 3D coordinates of the complex. These and other factors make it difficult to use even 3D information in developing machine learning models, much less 1D and 2D descriptors.
Data
Machine learning applied to facial recognition works well but that’s because we have a lot of data to train on; the universe of potential options is big but circumscribed; and substantial investments have been made in the technology by companies such as Apple, Facebook and Google. For example, the MegaFace algorithm, similar to that used in the new iPhone, is designed to accurately find the best matching face amongst ~7 billion people currently on planet earth. MegaFace performs well because it is trained on 1 million photos comprising about 690,000 different individuals. In the realm of drug discovery, things are quite a bit more challenging in this regard.
First, chemical space is really huge. To misquote Douglas Adams:
“[Chemical] space is big. You just won’t believe how vastly, hugely, mind-bogglingly big it is. I mean, you may think it’s a long way down the road to the chemist’s, but that’s just peanuts to space.”
Computational estimates suggest that the actual number of small molecules could be 1060, of which less than 1012 molecules (1,000 billion) have been made by academia and industry, a ratio similar to a drop of water in all the earth’s oceans. Our ability to appropriately describe and explore chemical space is currently highly limited.
Second, the cost of generating robust experimental data that a computer can make sense of is frustratingly slow and expensive. Well-resourced pharma drug discovery teams might make a thousand or so compounds en route to discovering a clinical candidate. Of these, perhaps a few hundred might be characterized across all the important properties such as potency, hERG, solubility etc. Producing more data isn’t currently economical, at least today. Compounds are expensive to make and the assays are bespoke and pricey. Together, this greatly limits the learning substrate that a computer can ingest and progress. Moreover, noise in the data from biological systems can confuse machine learning algorithms. It’s a surprise that machine learning works at all in chemistry but well-curated datasets for molecules that are similar in structure can work well.
Algorithms
Machine learning is a big topic for tech companies and the good news is that when it comes to the fundamental machine learning algorithms, we can apply much of this innovation in life sciences. TensorFlow developed by Google Brain, the deep learning research team at Google, offers an open source platform for anyone to use. Beyond that, there’s another ten options available for you to choose from. Bottom line, the machine learning algorithms themselves are not currently a bottleneck or a competitive advantage.
In contrast, developing algorithms that deliver realistic representations of nature are challenging and represent a big opportunity. This is because living in a 3D, dynamic, thermodynamic world can become very expensive. It can be challenging to obtain structures of your protein via X-ray and, even with a structure, allowing the protein to relax and move is computationally demanding.
The future
What is going to cause a step change improvement over the next 10 years? As we look at the constraints listed above, I am particularly excited by the following:
Better representations of nature: breakthroughs in image processing leveraged how the brain works, which makes sense because one of the key things done by the brain is to process visual information. Likewise, advances in human aviation required us to understand how birds fly. Similarly, if we can create algorithms that more realistically describe the fundamental nature of protein ligand binding, we will likely be able to better predict the phenomenon. A step forward here is Schrödinger’s industrialization of free energy perturbation (FEP) as is the Atomwise AtomNet Technology.
FEP can calculate how tightly a molecule binds to a protein. Since it is based solely on the rules of physics, its predictions correlate well with experiment, even in the case of Molecule A above. FEP offers a similar breakthrough for chemistry that Convolutional Neural Networks served for image processing. As molecular dynamics becomes cheaper and more high resolution structural data are available from approaches such as cryo-electron microscopy (cryo-EM), chemistry is going to follow a similar path of progress as image analysis.
Robust cost-effective data: good chemistry machine learning models require robust data on a broad set of molecules. There are two barriers to unlock: (i) making molecules is still an expensive, artisanal undertaking, at least for the more interesting compounds; and (ii) biological assays do not scale well and noise in the data can confound many machine learning algorithms. Over a 10-year timeframe, I think we’ll see approaches for on-demand chemistry as proposed by the DARPA ‘Make It’ program. New methodologies are being developed for making molecules that are gentler and more suitable for miniaturization, a trend that could begin to address the chemistry bottleneck.
One further observation is the fact that, in many situations, FEP described above does a good job of predicting potency of structurally related molecules. Hence, it is possible to use calculated data in lieu of experimental data to train a machine learning model. FEP currently costs around $3-150 computing cost per compound depending on the licensing model so it can be comparable to running a real assay. As costs come down, we may see FEP used broadly especially where the underlying chemistry is challenging e.g. macrocycles. Enabling this trend are significant increases in available computing power provided by graphics processing units (GPU’s), which incidentally are also used for bitcoin mining.
Accessing more chemical space: it is well known that computers can be trained to parse text to derive meaning and context using natural language processing (NLP) algorithms. Computers can also be trained to understand the grammar of chemistry. An elegant application of this approach is to (i) train a machine learning model using the rigorous FEP method and then (ii) evaluate billions of molecules generated by an algorithm that knows the rules of chemistry. The latter is important since it is pointless for a computer to suggest a molecule that’s physically impossible or cannot be made. The output of such approaches would then allow a much smaller number of molecules to be made.
Companies such as Schrödinger and Exscentia are pioneering such approaches and it is exciting to see meaningful deals with pharma that recognize the potential of these approaches.
Bringing diverse skill sets together: Of course, the barriers to the successful application of machine learning in chemistry are not all technology related. There are very real considerations around how to integrate machine learning skill sets into existing pharma companies. In addition, LifeSciVC, has noted that bottlenecks exist in drug discovery that are not currently addressed well by machine learning e.g. biology. There also exist cultural barriers to bringing together the classical Pharma MD, PhD skillset with the millennial-dominated Silicon Valley mentality.
A flurry of companies such as Benevolent.ai have sprung up recently built from the ground up around the concept of artificial intelligence. While there is definitely a bandwagon effect – 127 startups claim to use artificial intelligence in drug discovery based on this recent blog post – these companies imagine that we can turn the drug discovery model on its head with a completely new way of doing drug discovery, just as the Agile movement replaced the standard software development model. I like the fresh inspiration that our tech colleagues bring to the table but the jury is still out on whether these organizations can outperform Big Pharma using similar sets of tools, albeit in the context of a traditional organizational structure.
***
To conclude, I believe that artificial intelligence is overhyped but it would be a mistake to dismiss the impact that AI is going to have on drug discovery in the long run. The genomics revolution shows us how big technology shifts impact our industry, but also how long it takes to transform this promise into product value. The incredible strides that have been made in image and speech recognition provides a blueprint for how things may play out in Pharma. It is then really a matter of when we will see the impact of AI in drug discovery, not if.