



This blog was written by Michael Gladstone, Principal at Atlas Venture, as part of the From The Trenches feature of LifeSciVC.
Biotech newsflow came out the gate a little slow in 2018 (though it appears it may be speeding up today). So here I will speak about five burgeoning areas likely to generate some big news this year, each finally ready to break out after extensive multi-decades-long R&D iteration.
These aren’t necessarily “2018’s Biggest Catalysts,” but instead they represent a basket of spaces that have special significance at the translational medicine stage, at least from my individual perspective as an early-stage therapeutics investor with a focus in oncology, immunology, and new modalities
ECHO-301, FIRST LAYER: A MOMENT OF RECKONING FOR IDO (OR IS IT?)
Called the “mother of all binaries” by Leerink, ECHO-301 is Incyte’s trial of pembrolizumab + epacadostat in advanced melanoma. As the first readout of a randomized trial for this highly valued class, some think this will finally “end” the debate over the IDO class, one way or another. But I actually think, regardless of this trial’s outcome, the IDO discussion is just getting started. Several important questions will likely remain quite open after this trial:
- Was ECHO-301’s patient population (PD-1 naïve melanoma) the optimal indication? To what extent does success (or failure) in this setting read across to other indications in different tumors and in checkpoint-experienced patient populations? Or patients stratified by according to PD-L1 status, immunoscore, or tumor mutational burden?
- Was this dose of this drug the test of the IDO mechanism? Did this dose of epacadostat provide optimal target engagement and pharmacodynamics in the right compartments (blood/tumor/lymph nodes) for us to consider “the IDO clinical hypothesis” satisfactorily tested? Or does epacadostat’s potentially incomplete pathway inhibition point to opportunities for compounds with different potency, specificity, or PK?
- Is IDO-selective inhibition the optimal mechanistic test of the tryptophan > kynurenine > AHR pathway? How do we know an IDO-selective inhibitor is an optimal lever for this pathway? Is there a more powerful opportunity for therapeutics that address the other kynurenine-generating enzyme (“TDO”) or other ligands/inducers of AHR (the receptor for IDO’s enzyme product), both of which an IDO-selective inhibitor can’t impact?
Regardless of ECHO-301’s outcome, these judgments will impact many IO pipeline decisions. If the trial is successful, these are some important avenues for competitors to potentially differentiate their products. And if the trial is unsuccessful, a lot of competitors will need to either hang up their IDO shoes or ensure their agents address those potential liabilities. Either way, I suspect we’re actually still in the early days, and it will take more data to adequately answer any of these fundamental questions.
ECHO-301, SECOND LAYER: IMPLICATIONS FOR ALL THE OTHER PD-1 COMBO TRIALS
The other aspect of ECHO-301 is how it may also impact non-IDO PD-1 combo programs, by changing the future interpretation/perception of uncontrolled early clinical data. If IDO’s rich preclinical target validation and epacadostat’s apparent PD-1 additivity (in single-arm, uncontrolled studies) does not translate into robust Phase 3 success, Pharmas and investors may apply steeper discounts to the results of other single-arm IO combo studies in PD-1 responsive diseases, and it will become harder to get away with cross-trial data comparisons like this. That would have major implications for the design of IO “proof-of-concept” studies, perhaps driving to earlier use of randomized, controlled study designs and/or increased focus on truly PD-1 non-responsive or refractory disease settings, where single-arm data can be more meaningful (as an example, see what TLR9 agonists have done in PD-1 refractory melanoma).
THE YEAR OF THE CYTOKINE
Cytokines are ultra-potent proteins that provide the immune system its marching orders. I mean it when I say they are “ultra-potent” – as two examples, both IL-2 and IL-12 are dosed at microgram quantities, and even at those doses they frequently provoke life-threatening immune toxicities. Nonetheless, when tolerated, these cytokines can mediate spectacular clinical results.
Thus, like getting my dog to take a bath, developing safer cytokine therapies is very technically challenging but important enough to warrant repeated efforts. Several programs in the cytokine arena are making hard-fought progress:
- CD122-biased IL-2 (oncology): High-dose IL-2 is an approved, albeit infrequently used, melanoma/RCC regimen. Several companies are finally making progress divorcing IL-2’s infamous toxicity from its anti-tumor effects, especially Nektar with NKTR-214, which has demonstrated striking early activity with PD-1 inhibition in a variety of “hot/warm” tumors. Other clinical-stage programs include Alkermes’s ALKS-4230 and Roche’s cytokine fusion proteins.
- CD25-biased IL-2 (autoimmunity): I’ve previously described the flipside of the IL-2 biology noted above, and its immune-regulatory effects via Treg-modulation. At least three Treg-selective IL-2 muteins are expected in the clinic this year (Celgene, Amgen, and Nektar/Lilly), so hopefully we learn more soon about this approach’s potential.
- IL-10 (oncology): as IL-2 demonstrates, immunology is full of apparent paradoxes. So while I admit I was initially skeptical that IL-10 (canonically an “anti-inflammatory” cytokine) could be an effective oncology drug, ARMO’s accruing data with PEG-IL-10 suggests a CD8+ T cell-powered immune effect that combines well with PD-1 inhibition.
The last decade has produced major insights in cytokine biology (my mid-2000’s undergraduate research on IL-2 and IL-21 is already woefully outdated). So now we’re just arriving at the moment where our understanding of the immunobiology is catching up to industry’s protein engineering capabilities. Thus I believe this space is ripe for additional breakthroughs in rendering cytokines as better drugs, both as recombinant proteins and incorporated into next-generation cell therapies.
CAR-T GOES MAINSTREAM
2017 delivered the first two CAR-T FDA approvals (Novartis and Gilead/Kite), additional impressive CD19 data from Juno, and stunning data targeting BCMA in myeloma (Bluebird and Nanjing Legend). New Pharmas are entering the cell therapy fray in real time (see J&J’s recent Nanjing Legend deal, Amgen’s program AMG 119, and Takeda’s partnership with GammaDelta Therapeutics), not to mention a newly emerging generation of upstarts (e.g., Unum Therapeutics, Obsidian Therapeutics, Torque, and others). In 2018, we’ll see the early commercial roll-out of the first of these therapies and the battle to cement (and maintain) leadership in this fragmented, rapidly evolving space.
I’m most interested in seeing whether this generation of CAR products can be readily transformed into an outpatient therapy, and whether next-gen products (“armored CARs” or genetically augmented TIL/NK cell therapy) can extend cell therapy’s reach into solid tumors, though we won’t close the book on either of those questions in 2018.
IL-1b & CANTOS: A STUBBORN DISEASE YIELDS A NOVEL SURROGATE BIOMARKER
Cardiovascular disease has long been a challenging destination for early-stage R&D dollars. True proof of concept often comes late in development, due to the need for large trials in heterogeneous populations with long follow-up to uncover the hard outcomes data usually required for approval and commercial uptake. But Novartis’s CANTOS atherosclerosis trial of canakinumab (anti-IL-1b mAb) may revolutionize this landscape for two reasons.
First, this is the first anti-inflammatory intervention to produce an outcomes benefit in a large randomized atherosclerosis trial. This helps validate the hypothesis that systemic inflammation contributes to cardiovascular disease, and it provides an orthogonal (non-lipid modifying) therapeutic option to supplement in this patient population.
Secondly, this trial provides a treatment-responsive biomarker candidate that appears predictive of outcomes. As shown in the chart below, reproduced from this Lancet paper (and eloquently reviewed here), a treatment-emergent reduction in the circulating inflammation biomarker CRP (measured at 3 months) foretold improved outcomes in a study with a 3.7 year (!) median follow-up. This is proportionally like my knowing 30 minutes into cooking an 8-hour pot roast whether the meal is more likely to turn out tender and delectable or leathery and awful; of course the latter is still an unfortunate result, but knowing it in advance gives you time to make other dinner plans.
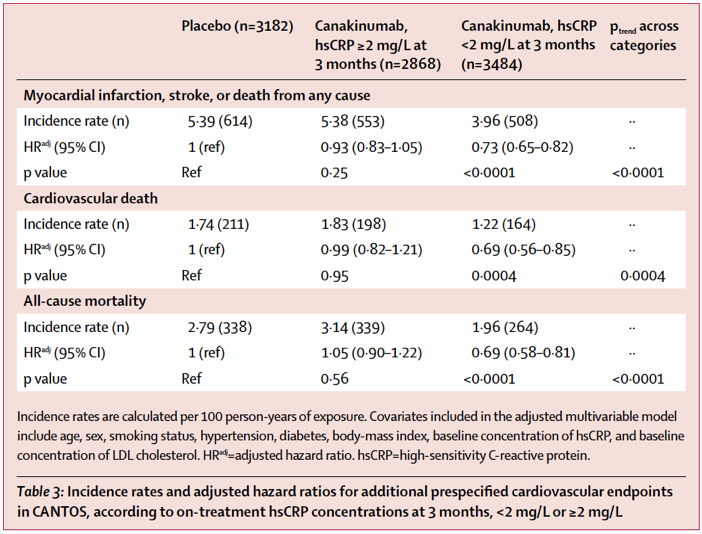
The CRP biomarker may help guide use of canakinumab by cardiologists, and it also may provide a useful and rapid surrogate that expedites and derisks development of other IL-1b-adjacent interventions like NLRP3 inhibitors, which will act upstream to reduce IL-1b secretion and other pathologic inflammatory actions.
Analogously, in the fibrosis field emerging imaging technologies and circulating biomarkers (e.g., MRE, C3M, pro-C3, collagen FSR) may similarly become increasingly validated as more rapid reflections of organ remodeling. This would pave the way for earlier demonstration of proof-of-concept for true antifibrotic therapies in NASH, IPF, etc. I hope similar opportunities present themselves in other areas where early proof-of-concept (on clinical endpoints) has been similarly hard to come by, such as neurodegeneration.
GENE THERAPY COMES OF AGE (FOR REAL THIS TIME)
This section title is stolen from an excellent recent Science Magazine review. Given the gene therapy field’s history of occasional false starts, there’s some pleasing irony that this review was preceded by a Nature review with the same title… published in 1992.
Nonetheless, this time is different (pardon the cliché, which I’ve used un-ironically in this setting). We now have robust clinical validation for AAV therapies in multiple diseases affecting confined compartments of post-mitotic cells (e.g., retina and CNS) and diseases addressed by relatively modest absolute quantities of “replacement protein” production by the liver (hemophilia A/B).
The key questions on the horizon for me:
- Can we convince patient, providers, and payors that these AAV programs have sufficiently favorable risk/benefit and pharmacoeconomic profiles to warrant widespread adoption and reimbursement?
- Will gene expression be sufficiently predictable and durable to safely, effectively, and permanently extend the clinical trial results to “the real world”?
- Can these (or other) vectors safely achieve adequate expression levels to address diseases with higher protein expression requirements than hemophilia?
- Can these (or other) vectors effectively and homogenously enough transduce other tissues (cardiac/skeletal muscle, lung, other CNS regions) to expand the pool of treatable diseases?
In 2018, we’ll come closer to addressing many of these questions for a variety of genetic medicine technologies:
- In the AAV field, we can look forward to maturing data on the lead programs in hemophilia, SMA, and Parkinson’s, and initial/additional data on the emerging set of clinical programs, including X-linked myotubular myopathy & Crigler Najjar, DMD, OTC deficiency, and Huntington’s.
- In the in vivo gene-editing space, Sangamo initiated their first in vivo gene editing clinical trials, using AAV delivery of zinc finger nucleases for permanently-integrated gene insertion in the liver. Other nuclease technologies (CRISPR, megaTALs) have similar ambitions in other diseases. Meanwhile, Logic Bio and Homology Medicines are nearing their first clinical trials, where they seek to accomplish AAV-assisted genome- editing/addition, but without using nucleases. Fast on their heels is the base-editing technology, which conducts site-specific DNA base exchange, possibly allowing correction of certain mutations with increased efficiency than HDR can so far muster.
- On the re-dosable gene therapy front, recently announced Generation Bio is developing a unique non-viral-encapsidated DNA vector that produces durable gene expression, permitting the step-wise titration and maintenance of the therapeutic effect over a patient’s lifetime and without the packaging constraints or manufacturing challenges imposed by AAV capsids.
- Hemoglobinopathy tipping point: This year we’ll see maturation of bluebird’s ex vivo lenti-HSC data in beta-thalassemia and sickle cell disease, as well as CRISPR Tx’s CTX001 (also for beta-thalassemia), which is the first company-sponsored CRISPR clinical trial.
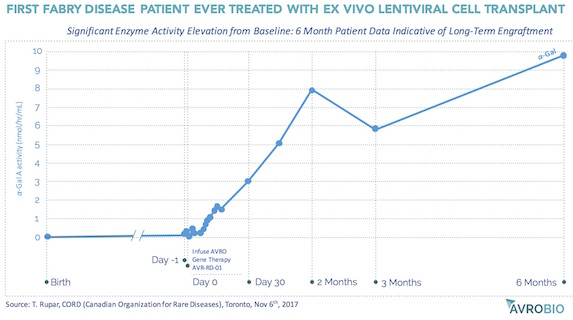
- Elsewhere in ex vivo HSC gene therapy, AvroBio and Orchard Therapeutics are two clinical-stage rising stars. Orchard has stunning data correcting ADA-SCID, and their early JPM-debuted results treating another rare inherited immune disorder (X-CGD) looks very promising as well. AvroBio uses autologous lentivirus-modified HSCs to provide durable, consistent replacement enzyme production throughout all of the body’s tissues and organs (since the gene-modified HSC-derived cells circulate through the blood and take up residence in tissues). They have disclosed compelling data from their first treated Fabry’s disease patient, and they plan to submit INDs/CTAs for two additional lysosomal storage disorders this year.
- mRNA therapy heads into non-vaccine trials: Cutting out the mRNA middleman entirely, Translate Bio (initially with its cystic fibrosis and OTC deficiency programs) and Moderna (VEGF-A and intratumoral OX40L) are ready to put mRNA therapy to the test in the clinic.
{kind=link}
In all, these technologies provide an unprecedented toolkit of genetic medicines to address the diversity of diseases (both rare and common, monogenic and complex) that have not yet been adequately addressed by more conventional modalities. It’s inconceivable to me that a one-size-fits-all approach will be get the job done, and so I look forward to seeing how each of these approaches play out in the clinic and settle in to addressing a diversity of new indications.
CONCLUSION
Hopefully I’ve convinced you that there’s a lot of interesting work afoot in large and small companies alike. And I’d be remiss not to point out two of the major contributing forces behind the programs I’ve highlighted here:
- Robust capital markets are facilitating and rewarding high-risk development: Most of the players mentioned in my gene therapy section are R&D stage companies, mostly less than 5 years old and many still private or only recently public. The major clinical-stage U.S.-based CAR-T players (Kite, Juno, and Bluebird) are/were all young entities that were able to capitalize risky, capital-intensive development programs that may change the way we treat cancer for many patients. Incyte and Nektar are revenue-generating companies that have been able to remain independent and thereby fund their respective R&D engines to discover and develop next-generation products that may not have been possible otherwise.
- The boldness of big companies: Novartis, Gilead, and Celgene investing in the global development of autologous cell therapies is not something most people would have predicted 5 years ago. The fact that big companies are committing major resources to CAR-T, AAV, gene editing, and 10,000-patient atherosclerosis trials of anti-inflammatory antibodies is a testament to the innovation instinct remaining very much alive in large biopharma companies.
There are a lot of things the biopharma and healthcare industries can do better. But there are also aspects of our industry that are working effectively as designed, to funnel resources toward talented people developing new medicines that may have been inconceivable a generation ago.