



Most conventional small molecule drugs engage directly with the engine of a protein’s function, aiming to often turning off an enzymatic activity associated with a disease. These drugs bind and compete against the natural substrate of the protein at what are known as orthosteric sites. But other pockets on a protein can also modulate function, and these sites commonly referred to as allosteric sites.
Nature commonly uses specific allosteric sites, distal to their active engines, to regulate the functional activity of proteins. Because of this potential powerful role, allostery has fascinated the pharmaceutical industry for decades as an approach that could open up otherwise challenging drug targets.
To date, the industry has made reasonable traction at making allosteric modulators for ion channels and transmembrane proteins on the surface of cells. Positive allosteric modulators (PAMs) and negative allosteric modulators (NAMs) are being exploited to regulate the activity of many cell types, but especially neurons, including the well-known benzodiazepine class of drugs. A broad range of PAMs and NAMs are being explored in clinical development today, including NMDA and SK2 channel programs.
This flavor of allostery has typically been uncovered via phenotypic cell-based screens looking for a downstream cellular effect; in many ways, these allosteric-site engagers are found by experimental “accident” with their binding sites only deconvoluted after the fact. These phenotypic screening methods to find allosteric modulators extends beyond just ion channels; agonists of enzyme activity in lysates and in cells can be uncovered in this HTS-enabled fashion, as occurred with our glucosylcerebrosidase (GCase) activator program at Lysosomal Therapeutics, now in Phase 1 for Parkinson’s Disease.
In light of the role of phenotypic screening in most cases, it’s no surprise then that most of the existing small molecule allosteric drugs were not created as a priori allosteric regulators of a protein, nor were they computationally-powered, protein structure-enabled programs (SBDD).
That said, there are a number of recent and notable examples of allosteric inhibitors discovered with significant input from SBDD, like Bcr-abl, Akt, HIF-2a, and SHP2 (here). At Nimbus, we also exploited this allosteric discovery strategy with both our acetyl CoA-carboxylase (ACC) and Tyk2 inhibition programs.
With ACC, Nimbus chose to go after an allosteric site on a regulatory domain of the enzyme, rather than the catalytic site where most of the industry had broken their pickaxe. Using SBDD, and enabled by a natural product that blocked the site, Nimbus discovered a potent inhibitor with attractive drug-like properties. After exciting early clinical data, Gilead acquired the program and is advancing it in NASH (here).
Building on these successes, Hotspot Therapeutics was founded to systematically approach the allosteric modulation of intracellular proteins by building a platform to exploit nature’s protein regulatory mechanisms. Today we are officially launching HotSpot out of seed-stage stealth mode with the closing of $45M Series A round of financing (http link).
Beginnings: It Starts With the Team
Hotspot was founded by CEO Jonathan Montagu and CSO Gerry Harriman. These two seasoned executives are very well known to Atlas, as they were two of our initial hires when I was acting CEO of Nimbus in 2010. Jonathan was the first CBO of Nimbus and Gerry led the successful discovery and early development of the ACC program. I had the pleasure of working with both of them very closely for a 4-6 years, and they left Nimbus in 2014 and 2016, respectively.

Jonathan Montagu and Gerry Harriman
The two of them began to noodle on the initial concept for HotSpot in late 2016, and we committed to seed the effort in December 2016, largely on the basis of two great serial entrepreneurs (who became Atlas EIRs) and a compelling concept paper. Other than our collective experience with SBDD-driven allostery, HotSpot had no specific lead matter around new targets or any new “rules” for allostery discovery at inception. It was a bet on a founding team with a great idea.
Gerry and Jonathan had a clear vision of what they wanted to build: a structurally-enabled product engine platform to a priori identify targets regulated by natural allostery and to discover novel drugs that exploit those mechanisms.
Shortly after seeding the startup in March 2017 with our friends at Sofinnova Partners (whom we had just worked closely with at Delinia), HotSpot assembled its core scientific team: Oscar Moradei drives medicinal chemistry (ex-Epizyme, Merck); Ara Aslanian heads up our in vitro pharmacology efforts (ex-Concert, Novartis); Peter Fekkes brings strong biophysical expertise as he leads the new targets effort (ex-H3, Novartis); Suresh Singh (ex-Vitae, Merck) has led our computational chemistry work; and, most recently, Christian Fritz joined to lead molecular pharmacology and translational biology (ex-Syros, Infinity).
With this solid team as a foundational element of our HotSpot investment thesis, what’s the nature of the opportunity?
Hotspots: privileged, natural allosteric regulatory sites
Drug discovery programs aimed at a protein’s catalytic or orthosteric site often face an uphill challenge in their quest for potent, selective, drug-like molecules. First, the inhibitor has to compete against natural substrate, which can reach very high concentrations inside of cells (e.g., ATP is mM in the cytosol); this creates high potency and target occupancy hurdles. Second, nature has largely conserved the catalytic engines that effect similar processes inside of cells (e.g., phosphorylating proteins via kinases), creating selectivity challenges that manifest as “off-target” concerns. Lastly, many substrates are charged or greasy (hydrophobic) which affects the character of these binding sites, biasing lead discovery towards chemistries with “un-drug-like” properties, challenging prospects for oral bioavailability and adequate pharmacokinetics.
Allosteric sites, in contrast, get around these liabilities by exploiting more natural regulatory contexts. Nature has evolved precise mechanisms for controlling protein function, often taking advantage of post-translational modifications (or PTMs, like phosphorylation, acetylation, methylation, etc). For example, many proteins are known to have large unstructured domains, far from their active sites, that get phosphorylated, inducing changes to protein structure through intramolecular phospho-regulatory binding sites. The same tertiary structure changes can happen with acetylation and other modifications.
The natural pockets where these PTMs fold or bind in unique ways create allosteric sites that are deemed regulatory “hotspots” – hence the name of the startup.
Drugging hotspots has some advantages over catalytic pockets. These sites don’t compete with abundant natural substrate. Because nature needs sensitive on/off mechanisms, these are typically very low affinity interactions. Further, they are often formed by unique protein sequences, which help solve the selectivity problem of conserved catalytic machinery. These sites also frequently have a more balanced drug-friendly character when it comes to their charge, hydrophobicity, and size. Lastly, many protein targets that are undruggable by conventional inhibitors can be targeted via these regulatory hotspots. All of this makes hotspot drug discovery incredibly compelling.
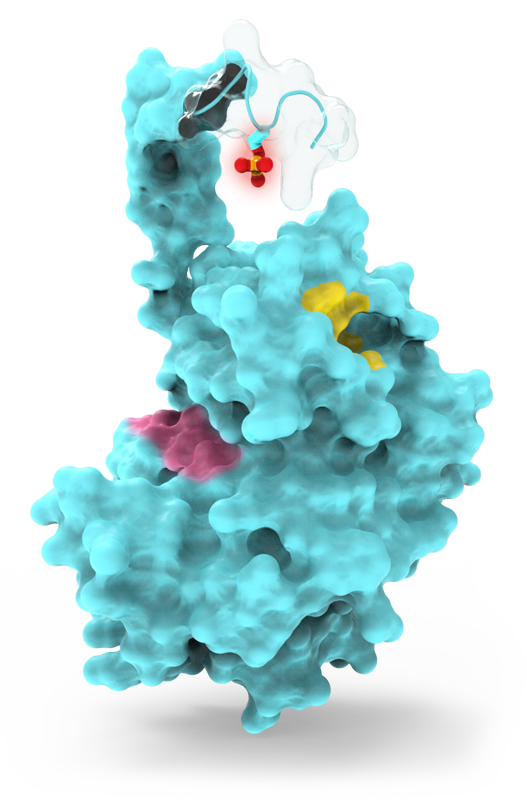 In addition, these hotspots are often superior to conventional forms of allostery: they are amendable to systematic SBDD approaches rather than phenotypic screens; they by definition have functional relevance vs other “cryptic” or random “computationally identified” protein pockets; and, the natural PTM ligand itself provides a starting point for a structural understanding of the site and initial chemical equity.
In addition, these hotspots are often superior to conventional forms of allostery: they are amendable to systematic SBDD approaches rather than phenotypic screens; they by definition have functional relevance vs other “cryptic” or random “computationally identified” protein pockets; and, the natural PTM ligand itself provides a starting point for a structural understanding of the site and initial chemical equity.
As for the scale of the opportunity, literally hundreds, if not thousands, of targets in the human proteome are amendable to hotspot-directed drug discovery. Our team has already characterized over 130 proteins with druggable allosteric sites.
HotSpot’s Platform And Product Engine.
At the heart of HotSpot’s discovery engine is its SpotFinderTM platform, a series of complementary technologies that enables pan-proteome mining of putative hotspots:
- site-mapping and virtual chemical probe techniques that prioritize potential hotspots that are predicted to bind chemical modifications (such as phosphate) and most amenable to small molecule drug discovery
- meta-level functional data relevant to regulatory hotspots from target and gene databases, thereby creating picture of the pharmacology one anticipates through small molecules
- natural language processing tools which scan scientific literature to identify proteins with potential regulatory hotspots where structure-function is understood in the scientific literature
Once SpotFinderTM has identified and characterized a high potential site/protein target, Hotspot’s discovery platform combines both virtual and physical library screening to identify hits. Based on insights into hotspot binding site configurations, a novel chemical library has been created comprising privileged scaffolds/chemotypes that are not typically found in conventional screening decks. To drive specific binding modes, Hotspot applies extensive protein engineering approaches that lock the target’s hotspot in the various conformations, including those likely to enlist the optimal type of pharmacology when bound. All of this is underpinned by state of the art computation drug design and lead optimization techniques.
HotSpot executes against this platform in a globally-distributed, virtually-integrated fashion, much like our experience at Nimbus and elsewhere. Truly proprietary elements remain purely in-house, including SpotFinderTM, chemical library design, computational approaches like virtual screening; outsourced components are physical medicinal chemistry, assay execution, crystallography, biophysics – all performed by top tier partners around the world. HotSpot is also at the vanguard of applying technology-enabled agile startup concepts to biotech, especially with Slack as their workhorse day-to-day productivity and conferencing platform (here).
HotSpot’s Pipeline & Future Plans
As with all new drug discovery engines, target selection is a critically important driver of eventual success (or failure). HotSpot spent much of its first year mining different target classes and specific proteins in order to characterize and prioritize what ones to go after. We continue to turn the crank of the machine to identify new hotspot-possessing targets to feed the drug discovery process, and will ramp efforts to widen our pipeline over time.
To date, we’ve only disclosed two of our active programs: PKC-theta for autoimmune disease and S6K for metabolism, broadly defined. Both of these hotspot inhibitor programs appear to offer unique and differentiated pharmacology versus traditional active site inhibitors, and we’re excited by their progress.
- PKC-theta is an immunokinase target offering potential to simultaneously stimulate regulatory T-cells (T-regs) while dampening effector T-cell function. Past industry attempts to drug the active ATP-binding site of PKC-theta had typically failed due to off-target effects and generally problematic chemotypes. We’ve uncovered a phosphoregulatory hotspot, previously unexploited by industry, and now have the first and only allosteric inhibitors of this kinase. These demonstrate all of the predicted attractive features (potency, selectivity, robust pharmacology), and the program is advancing into lead optimization.
- S6K is a metabolic enzyme that plays pivotal role in a cell’s sensitivity to insulin signaling and changes in energy metabolism. Work by Novartis showed that knocking out S6K1 in mice restores insulin sensitivity in mice on a high-fat diet, while increasing the number/size of mitochondria. These mice were jokingly called ‘flash’ mice due to their ability to run seemingly forever. Further, S6K appears to control the metabolic energetics of cell fate, such as regulating Th17 differentiation. Because of this role, inhibiting S6K offers important applications in a range of metabolic diseases e.g. NASH, diabetes, obesity, etc… However, the ATP/active site has never delivered selective, potent molecules with the right biodistribution properties. Using our SpotFinderTM platform, we have found a regulatory hotspot on S6 and already identified the first and only allosteric inhibitors, with highly differentiated properties (including selectivity) versus traditional active-site binders.
The rapid progress of these two programs in less than 12-months, as well as further undisclosed target activity, gives us solid confidence that hotspot-directed drug discovery will be a rich vein to mine for unique pharmacology – and this triggered our keen interest in powering up the story with a significant Series A financing.
As is often the case, a critical input to scaling this platform will be further access to both capital and expertise over time. To that end, we’re open to early discovery stage collaborations with partners where there’s a good fit and desire to unravel nature’s regulatory code on specific programs, as well as to long-term patient capital looking to help scale a big story.
Allosteric R&D is one of the most exciting areas of drug discovery today – but HotSpot is already turning up the heat even more.