



This blog was written by Adam Rosenberg, CEO of Rodin Therapeutics, as part of the From The Trenches feature of LifeSciVC.
After decades of challenges in neuroscience research and development, there is new hope that we can design better clinical studies with a higher probability of success. A key driver of this optimism: Advances in disease-relevant fluid and neuroimaging biomarkers.
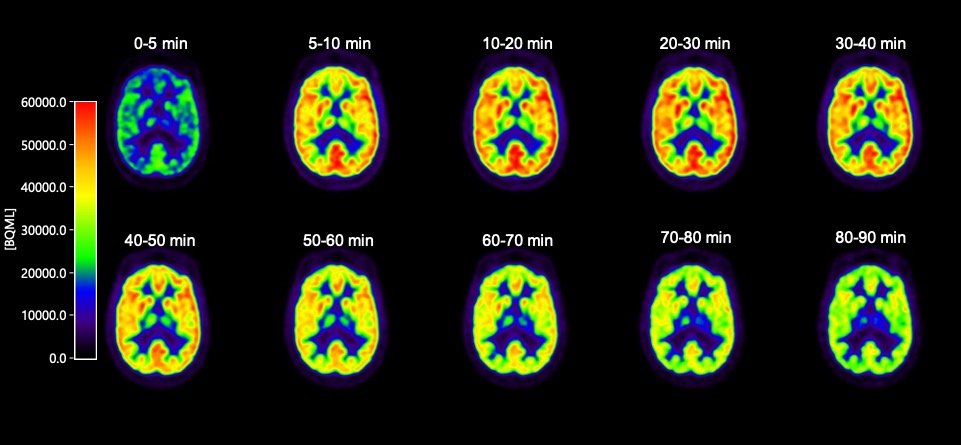
The SV2A PET tracer [11C]UCB-J is one such new, exciting neuroimaging tool. SV2A is a synaptic vesicle protein, located in the presynaptic terminals of neurons, and is a compelling biomarker for synaptic density. It is well-established that regardless of upstream pathobiology (eg, protein aggregation or the like), synaptic dysfunction is a key driver of symptoms across multiple brain diseases, and that synaptic damage and loss correlates with disease progression.
Historically, research on human synaptic loss has been done post-mortem. Now, with [11C]UCB-J, we have the ability to look inside living brains to assess synaptic density. To be sure, these are early days for this tracer; considerable work remains to be done. Yet the potential is huge, for both diagnostic and drug development purposes.
The neuroscience research community seems to be embracing the potential here and avoiding the winner-take-all sectarianism that so often plagues the field. Everyone we’ve spoken with who is involved in SV2A neuroimaging research – from both academia and industry – has been open and collaborative. This includes scientists from UCB, Yale and other organizations at the forefront of [11C]UCB-J clinical research.
In this spirit of open collaboration, this week we announced the initiation of a non-therapeutic study testing [11C]UCB-J to further assess its usefulness as a marker of synaptic density. Below, I’ll share a bit more about this clinical study, and how it fits within Rodin’s strategy and broader translational principles.
NON-THERAPEUTIC STUDY
Our non-therapeutic study of [11C]UCB-J is being conducted at two leading neurology centers in the Netherlands, in both healthy volunteers and Alzheimer’s patients. This study allows us to scan the same person twice, at day zero and day 28 (the same dosing duration as our upcoming Phase 1b in 2019), and thus assess scan-rescan variability over this period. These data will be critical as we assess study power and data reliability.
In addition, the practical operational benefits from such a non-therapeutic study cannot be understated; we’ve already learned a lot about the tracer’s chemistry and real-world performance, critical when incorporating an exciting but still novel translational tool into early studies. This study has also helped us develop a close working relationship with the centers that will participate in our Phase 1b study in 2019.
For a venture-backed company like Rodin to make a meaningful, seven-figure investment by sponsoring a clinical study of a generally-available imaging agent – a study that does not include our proprietary compounds – is a strong testament to our belief in the potential of the technology. This potential extends to our mechanism and others, and also broadly across synaptopathies (i.e., neurodegenerative, psychiatric and neurodevelopmental indications with symptoms driven by synaptic dysfunction). Indeed, an exciting feature of [11C]UCB-J clinical research is that the tracer is already being studied in multiple synaptopathies, including Alzheimer’s disease, Parkinson’s disease, schizophrenia and major depressive disorder.
To continue to advance SV2A neuroimaging research, we plan to work with our collaborators to publish the results of this non-therapeutic study as soon as possible after completion.
TRANSLATIONAL PRINCIPLES, AND RODIN’S STRATEGY
Translational strategies are not one-size-fits-all; different mechanisms require different approaches. The details that follow are Rodin-specific, but our hope is that by being transparent, we can at least prompt discussion and help provide a roadmap for others to compare their approaches. While our novel compounds are certainly proprietary, our translational strategy is not.
In my January 2017 post, I introduced our emphasis on synaptic resilience (here). This leads us in the direction of looking at markers of synaptic density and synaptogenesis, along with those of neuronal injury; a neuroinflammation or mitochondrial dysfunction approach would look different.
That said, there are key translational principles in early clinical studies that should hold across all mechanisms. Among them: measuring target engagement and pharmacodynamic (PD) activity, before undertaking more expensive Phase 2 trials.
As with any typical Phase 1 study, we plan to look at safety and pharmacokinetic (PK) measures (both central and peripheral) in single-ascending and multiple-ascending dose studies in healthy volunteers. In our case, we’ll also look at histone acetylation levels for a read on target engagement.
Our Phase 1b study will be in Alzheimer’s patients, and will be focused on confirmatory safety and PK measures , as well as proof of mechanism via biomarkers of synaptic density and neuronal injury (such as SV2A with [11C]UCB-J, and other fluid biomarkers such as SNAP25, PSD95, BDNF, neurogranin and neurofilament light chain).
We believe that these PD markers will provide a proper window to view potential drug response. They were also chosen in part because our compounds do not directly bind to them. Rather, drug-induced modulation of SV2A, BDNF or the like would be seen as the downstream molecular, structural and functional consequences of our therapy. These are the same principles we utilize when assessing preclinical activity, thereby reducing the leap of faith often required, eg when moving into the clinic based on rodent behavioral observations.
So, by assessing target engagement through histone acetylation, and PD effect primarily through synaptic biomarkers, we believe that after Phase 1b we’ll have a good early sense of whether our drug is safe and doing what we hope it will do at appropriate doses.
Further, [11C]UCB-J should allow us to directly measure synaptic density in specific brain regions. This is impossible with peripheral and CSF markers. Especially for a mechanism like ours that may have utility across a variety of synaptopathies, this could be highly valuable as we hopefully move into Phase 2.
***
Like other companies pressing ahead in neuroscience – including many who we look forward to speaking with at next week’s CTAD conference in Barcelona – we’ve tried to build a therapeutic thesis where we can learn from prior programs that failed to reach desired endpoints or outcomes. We’re also exploring new tools and patient enrichment approaches to drive our strategy in a way that is specifically tailored to our mechanism.
Will it work? What we can say for sure is that in Phase 1, we should learn whether our lead compound is safe and active in ways we desire in the brain. While no guarantee of success in later clinical studies (or certainly even in Phase 1), at minimum this approach allows us to make data-driven decisions on go/no-go, study design, choice of indications and endpoints.
There are many broader efforts to enhance translational collaboration and data-sharing, at the NIH level and elsewhere. By being transparent in our efforts as we move into the clinic, we hope that we’ll continue to enhance the collaborative approach shown by the community of researchers working on the SV2A PET ligand. While proprietary molecules are the lifeblood of biotech companies, translational data should be viewed as more open-source, especially given the unique challenges in neuroscience.
We’re looking forward to sharing the results of our non-therapeutic study as a first step in this direction.