



By Abbas Kazimi, CEO of Nimbus Therapeutics, as part of the From The Trenches feature of LifeSciVC
The Japanese Zen term shoshin means “beginner’s mind”. It’s the discipline of approaching familiar territory with fresh eyes and openness to see what may have changed. For an expert, this is not easy, because expertise tempts you to apply tried and true assumptions to new ideas.
Drug discovery may need a little more shoshin right now.
The pace of progress at the intersection of AI and medicine has been remarkable. New scientific workbenches, evolving protein-design systems, broad generative chemistry platforms, and increasingly capable coding agents have fueled large financing rounds, high-profile appointments, and a steady news cycle of ambitious predictions.
Yet for those of us who have spent our careers discovering and developing medicines, the honest reaction is complicated. There is excitement and curiosity, but also a quieter set of questions:
How much of this is hype? How is this all getting financed? Is AI truly new, or a more compelling description of computational approaches we have used for years? Do we understand why drug candidates fail well enough to know where AI can help? And, where are the medicines?
From my perspective as a drug hunter, the question is no longer whether AI can augment drug discovery. The question is what prevents us from effectively accessing these tools, learning to use them well, and applying them to scientific questions that matter.
A More Grounded AI Moment
Anthropic’s recent launch of Claude Science is an important signal of where the field is heading. The ambition is no longer AI as a chatbot that summarizes literature, but AI as an active participant in scientific work that could change how researchers search, analyze, code, model, and collaborate.
Across the industry, leading pharmaceutical companies are making significant investments to embed AI into the fabric of drug discovery. Bristol Myers Squibb recently announced plans to build what it describes as the most powerful AI factory in life sciences with NVIDIA, following Eli Lilly’s own investment in dedicated AI infrastructure and deployment. These investments are not just larger computing clusters. They reflect a recognition that AI infrastructure, secure access to proprietary scientific data, and the ability to deploy advanced models at enterprise scale are becoming strategic capabilities for modern pharmaceutical R&D.
But the path from better models to better medicines is not automatic. Derek Lowe, the medicinal chemist behind the long-running In the Pipeline blog, put it most memorably. Responding to Intel’s Andy Grove, who argued that drug discovery should take a page from the rapid acceleration of the semiconductor field, Lowe retorted that biology is not a system we have built from the ground up with fully understood governing rules. It is the product of billions of years of evolution. Better computation can help us navigate that complexity, but it does not make the complexity disappear.
That same tension appears in recent comments from Anthropic CEO Dario Amodei and Nobel laureate Jennifer Doudna, co-discoverer of CRISPR-Cas9. Amodei has been explicit that the impact of AI will depend on researchers learning to use it and organizations changing how they work. Doudna has cautioned against assuming that AI will replace the human ingenuity behind transformative discovery. Both perspectives lead to the same conclusion: more capable AI will not make biology any less difficult. It may accelerate discovery, but it will not remove the need for experimentation or judgment.
Holding those two ideas together, with confidence in AI’s potential and humility about what biology has yet to yield, may be one of the most important leadership challenges of this moment.
Different Roles in the AI-Enabled Drug Ecosystem
As powerful AI models become broadly available, what will differentiate one drug discovery organization from another?
The current conversation often groups very different organizations under the broad label of “AI drug discovery.” The boundaries are not perfectly clean, and some companies span more than one role, but I find it useful to distinguish among three parts of the ecosystem:
Tool builders create the models, software, predictive systems, and computational platforms that hopefully expand what drug discovery teams can do. Companies such as Isomorphic Labs and Chai Discovery are aiming to create tangible value by advancing the foundational models and platforms that enable AI-driven drug discovery.
Drug discovery companies, Nimbus included, integrate biology, chemistry, computation, experimentation, translational science, and portfolio judgment to create differentiated molecules and advance them into early clinical development.
Development and commercialization companies translate promising molecules into medicines through late-stage trials, regulatory approval, manufacturing, and patient access. This is where large pharmaceutical companies have historically excelled.
Nimbus is fundamentally a drug discovery company.
Throughout Nimbus’s history, our strategy has never been to build novel technologies ourselves. It has been to identify the technologies that create the most scientific leverage, integrate them fully into our discovery engine, and build proprietary capabilities only where they create lasting advantage.
We have applied that philosophy for years from computational chemistry and physics-based molecular design to machine learning, coding agents, scientific assistants, and connected workflows. Our partnership with AWS follows the same principle: rather than building foundational AI infrastructure of our own, the collaboration gives our scientists a secure, scalable environment to develop and deploy AI capabilities across active discovery programs.
The Advantage Is Learning Speed
Infrastructure enables discovery. It does not, by itself, differentiate discovery. When tools become broadly available, the advantage will shift from the tools themselves to how quickly an organization turns those tools into validated learning and better scientific decisions.
The speed that matters is the learning cycle:
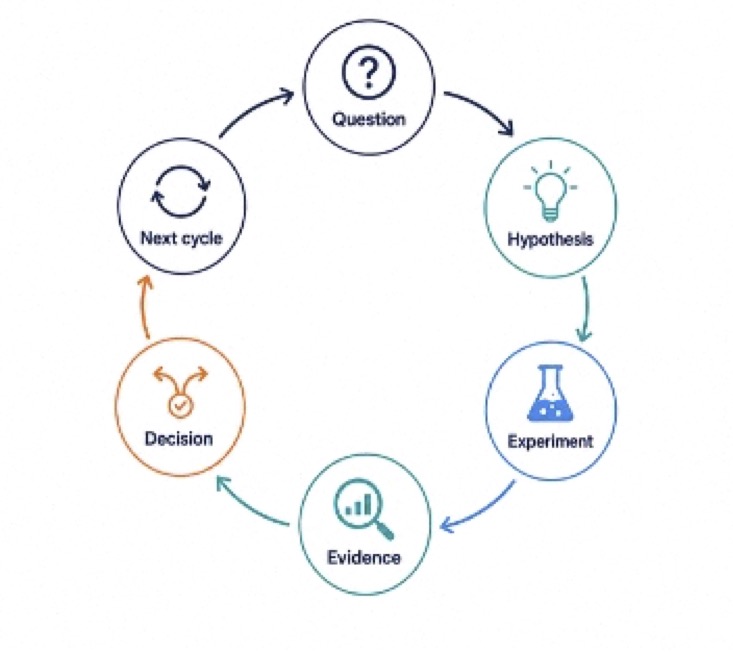 AI can compress that cycle by moving teams more quickly from question to evidence to decision. However, the advantage isn’t simply speed. It’s the speed of validated learning and every output still has to survive contact with the bench and the clinic. Without judgment, speed can simply generate more noise. Used with discipline, AI can help teams learn earlier, stop sooner, and advance better molecules.
AI can compress that cycle by moving teams more quickly from question to evidence to decision. However, the advantage isn’t simply speed. It’s the speed of validated learning and every output still has to survive contact with the bench and the clinic. Without judgment, speed can simply generate more noise. Used with discipline, AI can help teams learn earlier, stop sooner, and advance better molecules.
Speed is also relative. Drug discovery typically represents only the first four years of a decade or more of R&D, while late-stage clinical development accounts for most of the overall timeline and cost.
Accelerating early discovery is valuable, but improving the quality of decisions and advancing better molecules into development may have an even greater impact by reducing the risks that dominate the later stages of R&D.
What It Looks Like in Practice
In an earlier LifeSciVC post, I wrote that owning the learning loop matters more than owning the lab. AI makes that argument even more relevant.
At Nimbus, these tools are extending what great scientists can accomplish by shortening the distance between a scientific question and validated learning. This is not aspiration; it’s happening across our programs now. AI is embedded across the learning loop, from ideation and design through prediction, experimentation, and decision-making.
In one active program, our team built and deployed a custom pKa model in nine days to resolve a hERG-related design liability. In another, coding agents helped rewrite and extend a large pharmacophore and molecular shape-matching codebase in about a week, compressing work that might previously have taken months.
Across the portfolio, predictive ADME and property models increasingly guide which compounds we synthesize. Physics-based methods and machine learning are being combined to explore differentiated chemical space. We are also integrating agentic interfaces directly into our discovery workflows, enabling scientists to bring together literature, experimental data, calculations, specialized software, proprietary models, and internal decision frameworks through a common interface. These computational chemistry agents do more than make individual tools easier to use but rather help orchestrate the full workflow between a scientific question, a calculation, an experiment, and the next decision.
Our path has been evolutionary rather than a single leap. We began by giving teams broad access to tools such as Claude and Perplexity and then expanded into literature, patents, competitive intelligence, coding, and scientific synthesis. We are now developing reusable, domain-specific agents for target assessment, computational chemistry, medicinal chemistry, and other high-value workflows.
This is the advantage of embedding AI into a drug discovery company. The technology is tested continuously against active programs where the tools and programs improve each other through experimental feedback and validation.
What Actually Matters
The industry will continue to debate what should be called AI-designed, AI-enabled, AI-native, computational, generative, or agentic. Some distinctions are useful, but none is the final measure. What matters is whether these tools help us ask sharper questions, expose failure earlier, and move the right molecules forward.
At Nimbus, we believe they can, and the tools we have already deployed are making an impact. Rather than forcing AI into the center of an existing workflow to validate its use, we are extending what great drug hunters do and strengthening the learning loop around them.
As AI capabilities become widely available, the winners will not be the organizations that generate the most output. They will be the ones that approach new technologies with shoshin to learn faster, and turn what they learn into medicines that matter for patients.
Many thanks for the contributions and editorial advice from Daniel Price (Vice President, Computational Chemistry & Structural Biology, Nimbus) Cindy Fung (Vice President, Corporate Affairs & Communications, Nimbus), Peter Tummino (President, Research & Development, Nimbus), and Bruce Booth, (Partner, Atlas Venture).